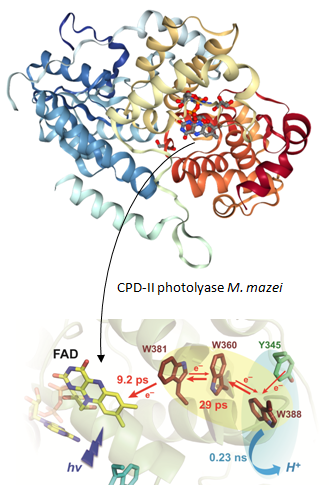
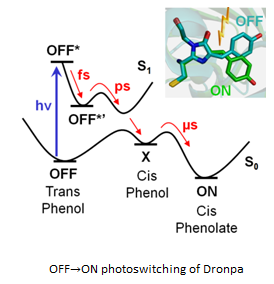
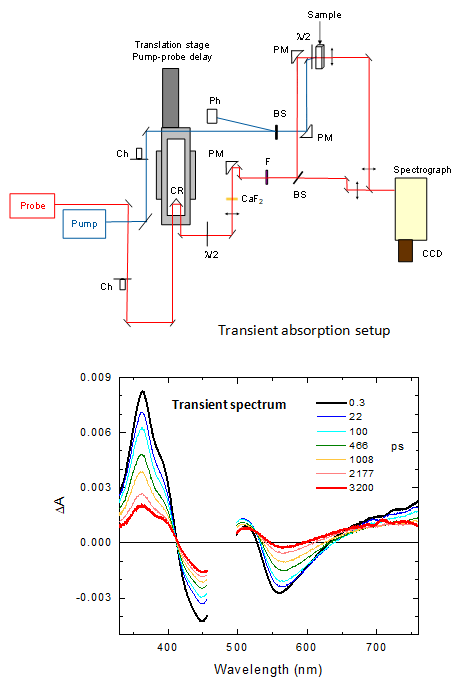
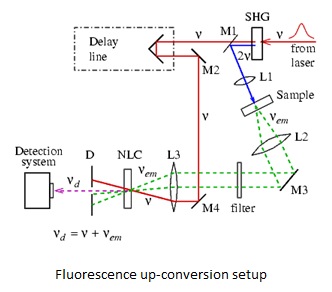
2002
|
A new analysis of the solvent-induced charge transfer in ADMA by subpicosecond spectroscopy Inproceedings M M Martin; P Plaza; P Changenet; A Siemiarczuk Douhal, A; Santamaría, J (Ed.): Vth International Conference on Femtochemistry, p. 280-288, World Scientific Publishing, 2002, ISBN: 0-444-51656-5. @inproceedings{RN92,
title = {A new analysis of the solvent-induced charge transfer in ADMA by subpicosecond spectroscopy},
author = {M M Martin and P Plaza and P Changenet and A Siemiarczuk},
editor = {A Douhal and J Santamar\'{i}a},
doi = {10.1016/b978-044451656-5/50081-5},
isbn = {0-444-51656-5},
year = {2002},
date = {2002-01-01},
booktitle = {Vth International Conference on Femtochemistry},
pages = {280-288},
publisher = {World Scientific Publishing},
series = {Femtochemistry and Femtobiology: Ultrafast Dynamics in Molecular Science},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
|
Excited-state relation dynamics of a PYP chromophore model in solution: Influence of the thioester group Article de journal P Changenet-Barret; A Espagne; N Katsonis; S Charier; J -B Baudin; L Jullien; P Plaza; M M Martin Chemical Physics Letters, 365 (3-4), p. 285–291, 2002. @article{Changenet-Barret:2002,
title = {Excited-state relation dynamics of a PYP chromophore model in solution: Influence of the thioester group},
author = {P Changenet-Barret and A Espagne and N Katsonis and S Charier and J -B Baudin and L Jullien and P Plaza and M M Martin},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0037109026&doi=10.1016%2fS0009-2614%2802%2901480-X&partnerID=40&md5=5fdfd4c614229bacad91377caee585a1},
doi = {10.1016/S0009-2614(02)01480-X},
year = {2002},
date = {2002-01-01},
journal = {Chemical Physics Letters},
volume = {365},
number = {3-4},
pages = {285--291},
abstract = {Cis-trans photoisomerization of a photoactive yellow protein chromophore model, the deprotonated trans S-phenyl thio-p-hydroxycinnamate, is studied in aqueous solution by subpicosecond transient absorption and gain spectroscopy. The excited-state deactivation is found to involve the formation, in 1.7 ps, of an intermediate state which decays in 2.8 ps. A persistent bleaching signal is observed at longer times indicating that the excited state not only relaxes to the ground state but also partly forms a stable photoproduct, possibly the cis isomer. This behavior is analogous to that of the native photoactive yellow protein. © 2002 Elsevier Science B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cis-trans photoisomerization of a photoactive yellow protein chromophore model, the deprotonated trans S-phenyl thio-p-hydroxycinnamate, is studied in aqueous solution by subpicosecond transient absorption and gain spectroscopy. The excited-state deactivation is found to involve the formation, in 1.7 ps, of an intermediate state which decays in 2.8 ps. A persistent bleaching signal is observed at longer times indicating that the excited state not only relaxes to the ground state but also partly forms a stable photoproduct, possibly the cis isomer. This behavior is analogous to that of the native photoactive yellow protein. © 2002 Elsevier Science B.V. All rights reserved. |
Multiple relaxation pathways in push-pull polyenes Article de journal D Laage; P Plaza; M Blanchard-Desce; M M Martin Photochemical and Photobiological Sciences, 1 (7), p. 526–535, 2002. @article{Laage:2002,
title = {Multiple relaxation pathways in push-pull polyenes},
author = {D Laage and P Plaza and M Blanchard-Desce and M M Martin},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-17444378861&doi=10.1039%2fb203201p&partnerID=40&md5=98848a0a1dd2e3c4a3b3b4729e644636},
doi = {10.1039/b203201p},
year = {2002},
date = {2002-01-01},
journal = {Photochemical and Photobiological Sciences},
volume = {1},
number = {7},
pages = {526--535},
abstract = {Subpicosecond absorption and gain spectroscopy are used to investigate the excited-state behavior of push-pull polyenes made of a diethylthiobarbituric acid electron-acceptor group and a dibutylaniline electron-donor group linked by a π-conjugated chain. Four polyenes of increasing length, ranging from n = 2 to 5 double bonds, are compared. The relaxation path and relaxation kinetics are studied in dioxane and in cyclohexane, a polar and a nonpolar solvent, respectively. In dioxane, the results provide evidence for the formation of an emissive transient state on an ultrashort time scale (2\textendash3 ps) attributed to a charge transfer (CT) state. The regular shift of the gain peak of this transient state with increase in the chain length (ca. 100 nm per added double bond) indicates that its structure is similar to that of a cyanine, i.e. with a fully conjugated polyenic chain. Its lifetime ranges from a few tens to a few hundreds of picoseconds depending on the chain length. When the number of double bonds increases from n = 2 to 3, the lifetime increases, then decreases continuously for longer chains. In cyclohexane, where the transient CT state is not formed, the decay of the initial excited state follows the same trend when the chain length increases but the lifetimes are shorter than that of the CT state in dioxane. In both solvents, the characterization of long-lived photoproducts by synchronizing two low repetition-rate subpicosecond laser systems demonstrates a change in the relaxation route as the chain length increases. Isomerization occurs for n = 2, whereas intersystem crossing to the triplet state occurs for n = 4. The change in the relaxation channel is observed for n = 3 in both solvents with however a solvent-dependent behavior. In dioxane, relaxation to the triplet state is already observed for n = 3, while an intermediate regime with a relaxation directly to the ground state is observed in cyclohexane. The photophysics of the studied push-pull polyenes is tentatively compared to that of polymethine cyanines and substituted carotenoids. © 2002 The Royal Society of Chemistry and Owner Societies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Subpicosecond absorption and gain spectroscopy are used to investigate the excited-state behavior of push-pull polyenes made of a diethylthiobarbituric acid electron-acceptor group and a dibutylaniline electron-donor group linked by a π-conjugated chain. Four polyenes of increasing length, ranging from n = 2 to 5 double bonds, are compared. The relaxation path and relaxation kinetics are studied in dioxane and in cyclohexane, a polar and a nonpolar solvent, respectively. In dioxane, the results provide evidence for the formation of an emissive transient state on an ultrashort time scale (2–3 ps) attributed to a charge transfer (CT) state. The regular shift of the gain peak of this transient state with increase in the chain length (ca. 100 nm per added double bond) indicates that its structure is similar to that of a cyanine, i.e. with a fully conjugated polyenic chain. Its lifetime ranges from a few tens to a few hundreds of picoseconds depending on the chain length. When the number of double bonds increases from n = 2 to 3, the lifetime increases, then decreases continuously for longer chains. In cyclohexane, where the transient CT state is not formed, the decay of the initial excited state follows the same trend when the chain length increases but the lifetimes are shorter than that of the CT state in dioxane. In both solvents, the characterization of long-lived photoproducts by synchronizing two low repetition-rate subpicosecond laser systems demonstrates a change in the relaxation route as the chain length increases. Isomerization occurs for n = 2, whereas intersystem crossing to the triplet state occurs for n = 4. The change in the relaxation channel is observed for n = 3 in both solvents with however a solvent-dependent behavior. In dioxane, relaxation to the triplet state is already observed for n = 3, while an intermediate regime with a relaxation directly to the ground state is observed in cyclohexane. The photophysics of the studied push-pull polyenes is tentatively compared to that of polymethine cyanines and substituted carotenoids. © 2002 The Royal Society of Chemistry and Owner Societies. |
Reversible bulk photorelease of strontium ion from a crown ether-linked merocyanine Article de journal P Plaza; I Leray; P Changenet-Barret; M M Martin; B Valeur ChemPhysChem, 3 (8), p. 668–674, 2002. @article{Plaza:2002,
title = {Reversible bulk photorelease of strontium ion from a crown ether-linked merocyanine},
author = {P Plaza and I Leray and P Changenet-Barret and M M Martin and B Valeur},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0037119287&doi=10.1002%2f1439-7641%2820020816%293%3a8%3c668%3a%3aAID-CPHC668%3e3.0.CO%3b2-G&partnerID=40&md5=e2d9cb7c397c5ade6faabcb1eb735f58},
doi = {10.1002/1439-7641(20020816)3:8<668::AID-CPHC668>3.0.CO;2-G},
year = {2002},
date = {2002-01-01},
journal = {ChemPhysChem},
volume = {3},
number = {8},
pages = {668--674},
abstract = {The use of a crown-ether-linked merocyanine, DCM-crown, for producing photoinduced long-lived and reversible concentration jumps of metalcations is reexamined. In this new investigation, the excited-state behavior of DCM-crown complexed with a strontium ion in acetonitrile is found to exhibit the same trends as those previously reported with calcium and lithium ions. However, some new features provide evidence for cation photorelease to the bulk. Subpicosecond transient absorption experiments confirm the initial fast photodisruption of the interaction between the ion and the crown, and the formation of a loose complex after intramolecular charge transfer within the chromophore. Two observed. Firstly, a continuous red shift of the gain spectrum is seen on the subnanosecond scale. It is assigned to the movement of the Sr2+ cation away from the chromophore, partly to the bulk of the solvent and partly towards the formation of an ultraloose complex with oxygen atoms of the crown. Secondly, a free-ligand-like absorption remains after the complete decay of the excited state. This band, which signals a total photorelease of the Sr2+ ion, disappears with a characteristic time of about 110 ns, attributed to the recomplexation of the crown in the ground state of the dye.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The use of a crown-ether-linked merocyanine, DCM-crown, for producing photoinduced long-lived and reversible concentration jumps of metalcations is reexamined. In this new investigation, the excited-state behavior of DCM-crown complexed with a strontium ion in acetonitrile is found to exhibit the same trends as those previously reported with calcium and lithium ions. However, some new features provide evidence for cation photorelease to the bulk. Subpicosecond transient absorption experiments confirm the initial fast photodisruption of the interaction between the ion and the crown, and the formation of a loose complex after intramolecular charge transfer within the chromophore. Two observed. Firstly, a continuous red shift of the gain spectrum is seen on the subnanosecond scale. It is assigned to the movement of the Sr2+ cation away from the chromophore, partly to the bulk of the solvent and partly towards the formation of an ultraloose complex with oxygen atoms of the crown. Secondly, a free-ligand-like absorption remains after the complete decay of the excited state. This band, which signals a total photorelease of the Sr2+ ion, disappears with a characteristic time of about 110 ns, attributed to the recomplexation of the crown in the ground state of the dye. |
UV-vis subpicosecond spectroscopy of 4-(9-anthryl)-N,N′-dimethylaniline in polar and nonpolar solvents: A two-dimensional view of the photodynamics Article de journal M M Martin; P Plaza; P Changenet-Barret; A Siemiarczuk Journal of Physical Chemistry A, 106 (10), p. 2351–2358, 2002. @article{Martin:2002,
title = {UV-vis subpicosecond spectroscopy of 4-(9-anthryl)-N,N′-dimethylaniline in polar and nonpolar solvents: A two-dimensional view of the photodynamics},
author = {M M Martin and P Plaza and P Changenet-Barret and A Siemiarczuk},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-0037075488&doi=10.1021%2fjp013107x&partnerID=40&md5=0302b1415347c0cd27d9e53261d1a8ca},
doi = {10.1021/jp013107x},
year = {2002},
date = {2002-01-01},
journal = {Journal of Physical Chemistry A},
volume = {106},
number = {10},
pages = {2351--2358},
abstract = {Transient absorption and gain spectra are reported for 4-(9-anthryl)-N,N′-dimethylaniline (ADMA) in polar and nonpolar solvents in the 330-780 nm wavelength range after excitation with a 500-fs laser at 370 nm. In acetonitrile, the initial transient spectra can be interpreted by the superposition of the dimethylaniline cation radical and the anthracene anion radical absorption bands, resulting from the well-known ultrafast photoinduced charge separation. In benzonitrile, similar transient spectra are reached 20-30 ps after excitation. In this solvent, as well as in alcohols, tetrahydrofuran and cyclohexane, the initial UV and visible absorption bands exhibit the spectral characteristics of the locally excited (LE) state. With increasing time, while these bands decay, the signature of the charge-transfer (CT) state appears and one observes a red shift of the region of minimum differential absorption, where gain is expected. These changes occur at a solvent-dependent rate, except in acetonitrile in which the spectra show little evolution because of our limited time resolution. Discrepancies with previously published models for ADMA photodynamics are discussed. In polar solvents except acetonitrile, a quasi-barrierless or slightly activated electron transfer is proposed to explain the long-time decay of the UV absorption band attributed to the LE state. On the other hand, inertial internal torsion around the bond linking the aniline and anthracene moieties in the LE state followed by solvation-induced charge separation similar to that in bianthryl is proposed to explain differences between acetonitrile and benzonitrile or other solvents in the UV range. A two-dimensional picture in which both internal torsion and solvation dynamics determine the excited-state reaction path is discussed, taking into account the initial proposal made by Mataga's group. In cyclohexane, in which solvation effects are not expected, the spectral changes in the picosecond time range are attributed to geometrical relaxation within the S1 state.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Transient absorption and gain spectra are reported for 4-(9-anthryl)-N,N′-dimethylaniline (ADMA) in polar and nonpolar solvents in the 330-780 nm wavelength range after excitation with a 500-fs laser at 370 nm. In acetonitrile, the initial transient spectra can be interpreted by the superposition of the dimethylaniline cation radical and the anthracene anion radical absorption bands, resulting from the well-known ultrafast photoinduced charge separation. In benzonitrile, similar transient spectra are reached 20-30 ps after excitation. In this solvent, as well as in alcohols, tetrahydrofuran and cyclohexane, the initial UV and visible absorption bands exhibit the spectral characteristics of the locally excited (LE) state. With increasing time, while these bands decay, the signature of the charge-transfer (CT) state appears and one observes a red shift of the region of minimum differential absorption, where gain is expected. These changes occur at a solvent-dependent rate, except in acetonitrile in which the spectra show little evolution because of our limited time resolution. Discrepancies with previously published models for ADMA photodynamics are discussed. In polar solvents except acetonitrile, a quasi-barrierless or slightly activated electron transfer is proposed to explain the long-time decay of the UV absorption band attributed to the LE state. On the other hand, inertial internal torsion around the bond linking the aniline and anthracene moieties in the LE state followed by solvation-induced charge separation similar to that in bianthryl is proposed to explain differences between acetonitrile and benzonitrile or other solvents in the UV range. A two-dimensional picture in which both internal torsion and solvation dynamics determine the excited-state reaction path is discussed, taking into account the initial proposal made by Mataga's group. In cyclohexane, in which solvation effects are not expected, the spectral changes in the picosecond time range are attributed to geometrical relaxation within the S1 state. |
2001
|
Femtosecond studies of intramolecular bond twisting in solution Inproceedings M Glasbeek; H Zhang; P Changenet; P Plaza; M M Martin; W Rettig Schryver, De F C; Feyter, De S; Schweitzer, G (Ed.): IVth International Conference on Femtochemistry, p. 417-430, Wiley-VCH, 2001. @inproceedings{RN99,
title = {Femtosecond studies of intramolecular bond twisting in solution},
author = {M Glasbeek and H Zhang and P Changenet and P Plaza and M M Martin and W Rettig},
editor = {F C De Schryver and S De Feyter and G Schweitzer},
year = {2001},
date = {2001-01-01},
booktitle = {IVth International Conference on Femtochemistry},
pages = {417-430},
publisher = {Wiley-VCH},
series = {Femtochemistry: With the Nobel Lecture of A. Zewail},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
|
Ionized Aminohydroxycarbene and Its Isomers: Relative Stability and Unimolecular Reactivity Article de journal G Bouchoux; A Espagne Chemical Physics Letters, 348 (3-4), p. 329-336, 2001, ISSN: 0009-2614. @article{RN55,
title = {Ionized Aminohydroxycarbene and Its Isomers: Relative Stability and Unimolecular Reactivity},
author = {G Bouchoux and A Espagne},
doi = {10.1016/s0009-2614(01)01117-4},
issn = {0009-2614},
year = {2001},
date = {2001-01-01},
journal = {Chemical Physics Letters},
volume = {348},
number = {3-4},
pages = {329-336},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Primary events in the photoactive yellow protein chromophore in solution Article de journal P Changenet-Barret; P Plaza; M M Martin Chemical Physics Letters, 336 (5-6), p. 439-444, 2001, ISSN: 0009-2614. @article{RN36,
title = {Primary events in the photoactive yellow protein chromophore in solution},
author = {P Changenet-Barret and P Plaza and M M Martin},
url = {<Go to ISI>://000167697200011},
issn = {0009-2614},
year = {2001},
date = {2001-01-01},
journal = {Chemical Physics Letters},
volume = {336},
number = {5-6},
pages = {439-444},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Primary photochemical processes in organic materials in solution Article de journal P Plaza; P Changenet-Barret; D Laage; M Martin Actualite Chimique, (2), p. 37–42, 2001. @article{Plaza:2001,
title = {Primary photochemical processes in organic materials in solution},
author = {P Plaza and P Changenet-Barret and D Laage and M Martin},
url = {https://www.scopus.com/record/display.uri?eid=2-s2.0-0035258302&origin=reflist&sort=plf-f&cite=2-s2.0-0035258302&src=s&imp=t&sid=5b7c48bdfaa5afb8c4c7e0247afd59c6&sot=cite&sdt=a&sl=0&recordRank=},
year = {2001},
date = {2001-01-01},
journal = {Actualite Chimique},
number = {2},
pages = {37--42},
abstract = {In this paper, we report on intramolecular electron transfer, distorsional relaxation or isomerization reactions in a few classes of commercial or recently engineered organic compounds in solution. We also show the role of the solvent polarity as well as solvent dynamics in the deactivation pathway and in the reaction kinetics. The reactions are probed in real time by pico-femtosecond transient absorption spectroscopy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In this paper, we report on intramolecular electron transfer, distorsional relaxation or isomerization reactions in a few classes of commercial or recently engineered organic compounds in solution. We also show the role of the solvent polarity as well as solvent dynamics in the deactivation pathway and in the reaction kinetics. The reactions are probed in real time by pico-femtosecond transient absorption spectroscopy. |
2000
|
Excited-State Dynamics in Polar Solvents of Push - Pull Polyenes Designed for Nonlinear Optics Article de journal P Plaza; D Laage; M M Martin; V Alain; M Blanchard-Desce; W H Thompson; J T Hynes Journal of Physical Chemistry A, 104 (11), p. 2396-2401, 2000, (cited By 27). @article{Plaza20002396,
title = {Excited-State Dynamics in Polar Solvents of Push - Pull Polyenes Designed for Nonlinear Optics},
author = {P Plaza and D Laage and M M Martin and V Alain and M {Blanchard-Desce} and W H Thompson and J T Hynes},
doi = {10.1021/jp992282z},
year = {2000},
date = {2000-01-01},
journal = {Journal of Physical Chemistry A},
volume = {104},
number = {11},
pages = {2396-2401},
abstract = {Subpicosecond spectroscopy of push - pull polyenes, previously designed to achieve large optical nonlinearities, reveals an isosbestic point in the time-resolved gain band in polar solvents. The compounds possess a diethylthiobarbituric acid electron-withdrawing group and a dibutylaniline electron-releasing group coupled by a $pi$-conjugated chain. The final gain peak exhibits a regular red shift with increasing chain length, whereas the ground-state absorption shift levels off. A fast photoinduced process toward a rigid, fully conjugated cyanine-like structure is proposed. This observation supports a recent proposal that a proper description of the excited-state dynamics of these push - pull polyenes requires more than two valence bond states.},
note = {cited By 27},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Subpicosecond spectroscopy of push - pull polyenes, previously designed to achieve large optical nonlinearities, reveals an isosbestic point in the time-resolved gain band in polar solvents. The compounds possess a diethylthiobarbituric acid electron-withdrawing group and a dibutylaniline electron-releasing group coupled by a $pi$-conjugated chain. The final gain peak exhibits a regular red shift with increasing chain length, whereas the ground-state absorption shift levels off. A fast photoinduced process toward a rigid, fully conjugated cyanine-like structure is proposed. This observation supports a recent proposal that a proper description of the excited-state dynamics of these push - pull polyenes requires more than two valence bond states. |