2008
|
Multiphotonic excitation and solvation dynamics effects on the femtosecond transient absorption of O-hexamethoxyhypericin Article de journal C Ley; J Brazard; F Lacombat; P Plaza; M M Martin; G A Kraus; J W Petrich Chemical Physics Letters, 457 (1-3), p. 82–86, 2008. @article{Ley:2008,
title = {Multiphotonic excitation and solvation dynamics effects on the femtosecond transient absorption of O-hexamethoxyhypericin},
author = {C Ley and J Brazard and F Lacombat and P Plaza and M M Martin and G A Kraus and J W Petrich},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-43549094927&doi=10.1016%2fj.cplett.2008.03.066&partnerID=40&md5=b5b213c38e5e9f6beff7cdfe2a3aa08b},
doi = {10.1016/j.cplett.2008.03.066},
year = {2008},
date = {2008-01-01},
journal = {Chemical Physics Letters},
volume = {457},
number = {1-3},
pages = {82--86},
abstract = {The hypothesis of a photoinduced intramolecular H-atom transfer in hypericin is reexamined by comparison with O-hexamethoxyhypericin, a derivative bearing no labile proton. In both cases, femtosecond transient absorption spectroscopy reveals an additional picosecond rising component in the kinetics under high excitation fluences. For the hexamethoxy derivative this effect is mixed with solvation dynamics but we succeeded in decoupling it. The delayed rise is ascribed to bi/multiphotonic excitation to upper excited states, followed by internal conversion to a hot S1 state that subsequently cools down. The present observations are not consistent with the intramolecular H-atom transfer interpretation. © 2008 Elsevier B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The hypothesis of a photoinduced intramolecular H-atom transfer in hypericin is reexamined by comparison with O-hexamethoxyhypericin, a derivative bearing no labile proton. In both cases, femtosecond transient absorption spectroscopy reveals an additional picosecond rising component in the kinetics under high excitation fluences. For the hexamethoxy derivative this effect is mixed with solvation dynamics but we succeeded in decoupling it. The delayed rise is ascribed to bi/multiphotonic excitation to upper excited states, followed by internal conversion to a hot S1 state that subsequently cools down. The present observations are not consistent with the intramolecular H-atom transfer interpretation. © 2008 Elsevier B.V. All rights reserved. |
Photoinduced intramolecular charge transfer in push-pull polyenes: Effects of solvation, electron-donor group, and polyenic chain length Article de journal W Akemann; D Laage; P Plaza; M M Martin; M Blanchard-Desce Journal of Physical Chemistry B, 112 (2), p. 358–368, 2008. @article{Akemann:2008,
title = {Photoinduced intramolecular charge transfer in push-pull polyenes: Effects of solvation, electron-donor group, and polyenic chain length},
author = {W Akemann and D Laage and P Plaza and M M Martin and M Blanchard-Desce},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-38749115416&doi=10.1021%2fjp075418z&partnerID=40&md5=bcc70b3649484de02d0eb2a114c93a58},
doi = {10.1021/jp075418z},
year = {2008},
date = {2008-01-01},
journal = {Journal of Physical Chemistry B},
volume = {112},
number = {2},
pages = {358--368},
abstract = {Subpicosecond absorption spectroscopy is used to characterize the primary photoinduced processes in a class of push-pull polyenes bearing a julolidine end group as the electron donor and a diethylthiobarbituric acid end group as the electron acceptor. The excited-state decay time and relaxation pathway have been studied for four polyenes of increasing chain length (n = 2-5 double bonds) in aprotic solvents of different solvation time, polarity, and viscosity. Intramolecular charge transfer (ICT) leading to a transient state of cyanine-like structure (fully conjugated with no bond length alternation) is observed in all polar solvents at a solvent dependent rate, but the reaction is not observed in cyclohexane, a nonpolar solvent. In polar solvents, the reaction time increases with the average solvation time but remains slightly larger, except in the viscous solvent triacetin. These facts are interpreted as an indication that both solvent reorganization and internal restructuring are involved in the ICT-state formation. The observed photodynamics resemble those we previously found for another class of polyenes bearing a dibutylaniline group as the donor, including a similar charge-transfer rate in spite of the larger electron donor character of the julolidine group. This observation brings further support to the proposal that an intramolecular coordinate is involved in the charge-transfer reaction, possibly a torsional motion of the donor end group. On the other hand, relaxation of the ICT state leads to cis-trans isomerization or crossing to the triplet state, depending on the length of the polyenic chain. In dioxane, tetrahydrofuran, and triacetin, the ICT state of the shorter chains (n = 2, 3) relaxes to the isomer with a viscosity-dependent rate, while that of the longer ones (n = 4, 5) leads to the triplet state with a viscosity-independent rate, as expected. In acetonitrile, the ICT-state lifetime is generally much shorter. A change from photoisomerization to intersystem crossing at n = 4 is also proposed in this solvent, but the formation of a photoproduct at n = 2 is not clear. In cyclohexane, where the ICT state is not formed, the relaxation pathway of the initially excited state is found to lead to an isomer for n = 2. As in polar solvents, a change to intersystem crossing at n = 4 is proposed. The direct relaxation to the ground state found at n = 3 for the series bearing a dibutylaniline group is not observed with the julolidine group. The results clearly illustrate that photoinduced reaction trajectories in push-pull polyenes are controlled by the static and dynamic properties of the solvent, the chemical nature and size of the end groups, and the conjugated-chain length and flexibility. © 2008 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Subpicosecond absorption spectroscopy is used to characterize the primary photoinduced processes in a class of push-pull polyenes bearing a julolidine end group as the electron donor and a diethylthiobarbituric acid end group as the electron acceptor. The excited-state decay time and relaxation pathway have been studied for four polyenes of increasing chain length (n = 2-5 double bonds) in aprotic solvents of different solvation time, polarity, and viscosity. Intramolecular charge transfer (ICT) leading to a transient state of cyanine-like structure (fully conjugated with no bond length alternation) is observed in all polar solvents at a solvent dependent rate, but the reaction is not observed in cyclohexane, a nonpolar solvent. In polar solvents, the reaction time increases with the average solvation time but remains slightly larger, except in the viscous solvent triacetin. These facts are interpreted as an indication that both solvent reorganization and internal restructuring are involved in the ICT-state formation. The observed photodynamics resemble those we previously found for another class of polyenes bearing a dibutylaniline group as the donor, including a similar charge-transfer rate in spite of the larger electron donor character of the julolidine group. This observation brings further support to the proposal that an intramolecular coordinate is involved in the charge-transfer reaction, possibly a torsional motion of the donor end group. On the other hand, relaxation of the ICT state leads to cis-trans isomerization or crossing to the triplet state, depending on the length of the polyenic chain. In dioxane, tetrahydrofuran, and triacetin, the ICT state of the shorter chains (n = 2, 3) relaxes to the isomer with a viscosity-dependent rate, while that of the longer ones (n = 4, 5) leads to the triplet state with a viscosity-independent rate, as expected. In acetonitrile, the ICT-state lifetime is generally much shorter. A change from photoisomerization to intersystem crossing at n = 4 is also proposed in this solvent, but the formation of a photoproduct at n = 2 is not clear. In cyclohexane, where the ICT state is not formed, the relaxation pathway of the initially excited state is found to lead to an isomer for n = 2. As in polar solvents, a change to intersystem crossing at n = 4 is proposed. The direct relaxation to the ground state found at n = 3 for the series bearing a dibutylaniline group is not observed with the julolidine group. The results clearly illustrate that photoinduced reaction trajectories in push-pull polyenes are controlled by the static and dynamic properties of the solvent, the chemical nature and size of the end groups, and the conjugated-chain length and flexibility. © 2008 American Chemical Society. |
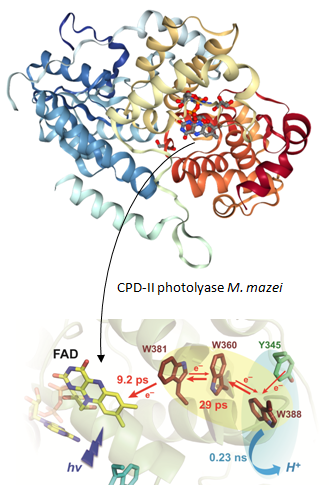
Polarized Transient Absorption to Resolve Electron Transfer between Tryptophans in DNA Photolyase Article de journal M Byrdin; S Villette; A Espagne; A P M Eker; K Brettel Journal of Physical Chemistry B, 112 (22), p. 6866-6871, 2008, ISSN: 1520-6106. @article{RN33b,
title = {Polarized Transient Absorption to Resolve Electron Transfer between Tryptophans in DNA Photolyase},
author = {M Byrdin and S Villette and A Espagne and A P M Eker and K Brettel},
doi = {10.1021/jp711435y},
issn = {1520-6106},
year = {2008},
date = {2008-01-01},
journal = {Journal of Physical Chemistry B},
volume = {112},
number = {22},
pages = {6866-6871},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Primary photoprocesses involved in the sensory protein for the photophobie response of Blepharisma japonicum Article de journal J Brazard; C Ley; F Lacombat; P Plaza; M M Martin; G Checcucci; F Lenci Journal of Physical Chemistry B, 112 (47), p. 15182–15194, 2008. @article{Brazard:2008,
title = {Primary photoprocesses involved in the sensory protein for the photophobie response of Blepharisma japonicum},
author = {J Brazard and C Ley and F Lacombat and P Plaza and M M Martin and G Checcucci and F Lenci},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-57449093150&doi=10.1021%2fjp805815e&partnerID=40&md5=e50debedfb440878c5edd474d3c3cd96},
doi = {10.1021/jp805815e},
year = {2008},
date = {2008-01-01},
journal = {Journal of Physical Chemistry B},
volume = {112},
number = {47},
pages = {15182--15194},
abstract = {We present new femtosecond transient-absorption and picosecond fluorescence experiments performed on OBIP, the oxyblepharismin-binding protein believed to trigger the Photophobie response of the ciliatc Blepharisma japonicum. The formerly identified heterogeneity of the sample is confirmed and rationalized in terms of two independent populations, called rOBlP and nrOBIP. The rOBIP population undergoes a fast photocycle restoring the initial ground state in less than 500 ps. Intermolecular electron transfer followed by electron recombination is identified as the excited-state decay route. The experimental results support the coexistence of the oxyblepharismin (OxyBP) radical cation signature with a stimulated-emission signal at all times of the evolution of the transient-absorption spectra. This observation is interpreted by an equilibrium being reached between the locally excited state and a charge-transfer state on the ground of a theory developed by Mataga and co-workers to explain the fluorescence quenching of aromatic hydrogen-bonded donor-acceptor pairs in nonpolar solvents. OxyBP is supposed to bind to an as yet unknown electron acceptor by a hydrogen-bond (HB) and the coordinate along which forward and backward electron transfer proceed is assumed to be the shift of the HB proton. The observed kinetic isotope effect supports this interpretation. Protein relaxation is finally proposed to accompany the whole process and give rise to the highly multiexponential observed dynamics. As previously reported, the fast photocycle of rOBIP can be interpreted as an efficient sunscreen mechanism that protects Blepharisma japonicum from continuous irradiation. The nrOBIP population, the transient-absorption of which strongly reminds that of free OxyBP in solution, might be proposed to actually trigger the Photophobie response of the organism through excited-state deprotonation of the chromophorc occurring in the nanosecond regime. Additional femtosecond transient-absorption spectra of OxyBP and perideprotonated OxyBP are also reported and used as a comparison basis to interpret the results on OBIP. © 2008 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We present new femtosecond transient-absorption and picosecond fluorescence experiments performed on OBIP, the oxyblepharismin-binding protein believed to trigger the Photophobie response of the ciliatc Blepharisma japonicum. The formerly identified heterogeneity of the sample is confirmed and rationalized in terms of two independent populations, called rOBlP and nrOBIP. The rOBIP population undergoes a fast photocycle restoring the initial ground state in less than 500 ps. Intermolecular electron transfer followed by electron recombination is identified as the excited-state decay route. The experimental results support the coexistence of the oxyblepharismin (OxyBP) radical cation signature with a stimulated-emission signal at all times of the evolution of the transient-absorption spectra. This observation is interpreted by an equilibrium being reached between the locally excited state and a charge-transfer state on the ground of a theory developed by Mataga and co-workers to explain the fluorescence quenching of aromatic hydrogen-bonded donor-acceptor pairs in nonpolar solvents. OxyBP is supposed to bind to an as yet unknown electron acceptor by a hydrogen-bond (HB) and the coordinate along which forward and backward electron transfer proceed is assumed to be the shift of the HB proton. The observed kinetic isotope effect supports this interpretation. Protein relaxation is finally proposed to accompany the whole process and give rise to the highly multiexponential observed dynamics. As previously reported, the fast photocycle of rOBIP can be interpreted as an efficient sunscreen mechanism that protects Blepharisma japonicum from continuous irradiation. The nrOBIP population, the transient-absorption of which strongly reminds that of free OxyBP in solution, might be proposed to actually trigger the Photophobie response of the organism through excited-state deprotonation of the chromophorc occurring in the nanosecond regime. Additional femtosecond transient-absorption spectra of OxyBP and perideprotonated OxyBP are also reported and used as a comparison basis to interpret the results on OBIP. © 2008 American Chemical Society. |
Real-time probing of fast photoinduced charge-transfer in electron donor-acceptor model compounds and in biological photosensors Article de journal M M Martin; P Plaza; P Changenet-Barret Polish Journal of Chemistry, 82 (4), p. 759-771, 2008, ISSN: 0137-5083. @article{RN58,
title = {Real-time probing of fast photoinduced charge-transfer in electron donor-acceptor model compounds and in biological photosensors},
author = {M M Martin and P Plaza and P Changenet-Barret},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-42649142889&partnerID=40&md5=93d132ea839d81faa18476dd4eaaf617},
issn = {0137-5083},
year = {2008},
date = {2008-01-01},
journal = {Polish Journal of Chemistry},
volume = {82},
number = {4},
pages = {759-771},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Steady-state and femtosecond photoinduced processes of blepharismins bound to alpha-crystallin Article de journal T Youssef; J Brazard; C Ley; F Lacombat; P Plaza; M M Martin; A Sgarbossa; G Checcucci; F Lenci Photochemical and Photobiological Sciences, 7 (7), p. 844–853, 2008. @article{Youssef:2008,
title = {Steady-state and femtosecond photoinduced processes of blepharismins bound to alpha-crystallin},
author = {T Youssef and J Brazard and C Ley and F Lacombat and P Plaza and M M Martin and A Sgarbossa and G Checcucci and F Lenci},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-46449089636&doi=10.1039%2fb800848e&partnerID=40&md5=948e794e195f413bde104725b45524da},
doi = {10.1039/b800848e},
year = {2008},
date = {2008-01-01},
journal = {Photochemical and Photobiological Sciences},
volume = {7},
number = {7},
pages = {844--853},
abstract = {The interaction of blepharismin (BP) and oxyblepharismin (OxyBP) with bovine alpha-crystallin (BAC) has been studied both by steady-state and femtosecond spectroscopy, with the aim of assessing the possible phototoxicity of these compounds toward the eye tissues. We showed that these pigments form with BAC potentially harmful ground-state complexes, the dissociation constants of which have been estimated to be 6 ± 2 μmol L-1 for OxyBP and 9 ± 4 μmol L-1 for BP. Irradiation with steady-state visible light of solutions of blepharismins in the presence of BAC proved to induce a quenching of both the pigment and the intrinsic protein fluorescences. These effects were tentatively rationalized in terms of structural changes of alpha-crystallin. On the other hand, femtosecond transient absorption spectroscopy was used to check the occurrence of any type I photoactivity of oxyblepharismin bound to alpha-crystallin. The existence of a particular type of fast photoinduced reaction, not observed in former studies with human serum albumin but present in the natural oxyblepharismin-binding protein, could here be evidenced but no specific reaction was observed during the first few nanoseconds after excitation. Partial denaturation of alpha-crystallin was however found to alter the excited-state behaviour of its complex with oxyblepharismin, making it partly resemble that of free oxyblepharismin in solution. © The Royal Society of Chemistry and Owner Societies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The interaction of blepharismin (BP) and oxyblepharismin (OxyBP) with bovine alpha-crystallin (BAC) has been studied both by steady-state and femtosecond spectroscopy, with the aim of assessing the possible phototoxicity of these compounds toward the eye tissues. We showed that these pigments form with BAC potentially harmful ground-state complexes, the dissociation constants of which have been estimated to be 6 ± 2 μmol L-1 for OxyBP and 9 ± 4 μmol L-1 for BP. Irradiation with steady-state visible light of solutions of blepharismins in the presence of BAC proved to induce a quenching of both the pigment and the intrinsic protein fluorescences. These effects were tentatively rationalized in terms of structural changes of alpha-crystallin. On the other hand, femtosecond transient absorption spectroscopy was used to check the occurrence of any type I photoactivity of oxyblepharismin bound to alpha-crystallin. The existence of a particular type of fast photoinduced reaction, not observed in former studies with human serum albumin but present in the natural oxyblepharismin-binding protein, could here be evidenced but no specific reaction was observed during the first few nanoseconds after excitation. Partial denaturation of alpha-crystallin was however found to alter the excited-state behaviour of its complex with oxyblepharismin, making it partly resemble that of free oxyblepharismin in solution. © The Royal Society of Chemistry and Owner Societies. |
Ultrafast photochemical processes: From the lab to natural photoactive systems Article de journal M M Martin; P Plaza; P Changenet-Barret; A Espagne; M Mahet; C Ley; F Lacombat Actualite Chimique, (320-321), p. 14–19, 2008. @article{Martin:2008,
title = {Ultrafast photochemical processes: From the lab to natural photoactive systems},
author = {M M Martin and P Plaza and P Changenet-Barret and A Espagne and M Mahet and C Ley and F Lacombat},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-48649089855&partnerID=40&md5=5dbe5748299ea91b50952e61834a11b9},
year = {2008},
date = {2008-01-01},
journal = {Actualite Chimique},
number = {320-321},
pages = {14--19},
abstract = {The UV-Vis subpicosecond absorption spectroscopy has been used to characterize and probe in real-time the primary photoinduced chemical processes in two photosensory proteins extracted from photomotile micro-organisms. This approach to the structure-function relationship of these photoactive proteins consists in comparing their photophysics to that of their chromophore isolated in solution, in order to evidence the specificity of the natural chromophore-protein complex. It has been shown that the photoisomerisation kinetic and quantum yield of the chromophore of photoactive yellow protein (PYP) are controlled by the properties of the protein nanospace in which it is embedded, and also that the oxyblepharism binding protein (OBIP) undergoes photoinduced picosecond reactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The UV-Vis subpicosecond absorption spectroscopy has been used to characterize and probe in real-time the primary photoinduced chemical processes in two photosensory proteins extracted from photomotile micro-organisms. This approach to the structure-function relationship of these photoactive proteins consists in comparing their photophysics to that of their chromophore isolated in solution, in order to evidence the specificity of the natural chromophore-protein complex. It has been shown that the photoisomerisation kinetic and quantum yield of the chromophore of photoactive yellow protein (PYP) are controlled by the properties of the protein nanospace in which it is embedded, and also that the oxyblepharism binding protein (OBIP) undergoes photoinduced picosecond reactions. |
2007
|
Cellular photoperception and photoactive proteins Article de journal M Martin; S Haacke; M Chergui; P Plaza; P Changenet-Barret; K Brettel; M Byrdin; J Y Bigot; L Guidoni; P Didier Actualité Chimique, 308-309 , p. 19-24, 2007, ISSN: 0151-9093. @article{RN54,
title = {Cellular photoperception and photoactive proteins},
author = {M Martin and S Haacke and M Chergui and P Plaza and P Changenet-Barret and K Brettel and M Byrdin and J Y Bigot and L Guidoni and P Didier},
url = {https://www.scopus.com/record/display.uri?eid=2-s2.0-34547297831&origin=resultslist&sort=plf-f&cite=2-s2.0-85005697420&src=s&imp=t&sid=4bf7c1307b95cbc645e86552103ccc27&sot=cite&sdt=a&sl=0&relpos=18&citeCnt=0&searchTerm=},
issn = {0151-9093},
year = {2007},
date = {2007-01-01},
journal = {Actualit\'{e} Chimique},
volume = {308-309},
pages = {19-24},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Photoinduced charge shift as the driving force for the excited-state relaxation of analogues of the Photoactive Yellow Protein chromophore in solution Article de journal A Espagne; P Changenet-Barret; J -B Baudin; P Plaza; M M Martin Journal of Photochemistry and Photobiology A: Chemistry, 185 (2-3), p. 245-252, 2007, ISSN: 1010-6030. @article{RN51,
title = {Photoinduced charge shift as the driving force for the excited-state relaxation of analogues of the Photoactive Yellow Protein chromophore in solution},
author = {A Espagne and P Changenet-Barret and J -B Baudin and P Plaza and M M Martin},
url = {<Go to ISI>://000243736600019},
doi = {10.1016/j.jphotochem.2006.06.016},
issn = {1010-6030},
year = {2007},
date = {2007-01-01},
journal = {Journal of Photochemistry and Photobiology A: Chemistry},
volume = {185},
number = {2-3},
pages = {245-252},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Primary photoprocesses in oxyblepharismin interacting with its native protein partner Article de journal M Mahet; P Plaza; M M Martin; G Checcucci; F Lenci Journal of Photochemistry and Photobiology A: Chemistry, 185 (2-3), p. 345–353, 2007. @article{Mahet:2007,
title = {Primary photoprocesses in oxyblepharismin interacting with its native protein partner},
author = {M Mahet and P Plaza and M M Martin and G Checcucci and F Lenci},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-33845674602&doi=10.1016%2fj.jphotochem.2006.06.030&partnerID=40&md5=292f9acdb239c0f7be41d6344b5be148},
doi = {10.1016/j.jphotochem.2006.06.030},
year = {2007},
date = {2007-01-01},
journal = {Journal of Photochemistry and Photobiology A: Chemistry},
volume = {185},
number = {2-3},
pages = {345--353},
abstract = {The primary photoprocesses in the photoreceptor for the step-up photophobic response of the light-adapted cells of Blepharisma japonicum (OBIP, OxyBlepharismin bInding Protein) have been studied by ultrafast UV-vis transient spectroscopy. The results are rationalized in terms of heterogeneity of the OBIP sample. Two independent classes of chromoprotein are proposed: a "reactive" species, which presents a specific 680-nm band decaying in 4 and 56 ps and a "non-reactive" one, which behaves like the free chromophore (OxyBP) in solution. A bimolecular photooxidation of OxyBP in the presence of 1,4-benzoquinone was performed to record the absorption spectrum of the OxyBP radical cation. Comparison with reactive OBIP suggests that an electron transfer could be involved in the primary photoprocesses of OBIP and possibly trigger the sensory transduction chain of B. japonicum. In addition, the specificity of the chromophore-protein interaction was investigated by studying the artificial complex that OxyBP forms with human serum albumin (HSA). OxyBP-HSA happens to be spectroscopically much closer to free OxyBP than to OBIP. This highlights the specific nature of the interaction between OxyBP and its native protein partner and further supports the proposal that OBIP is the actual photoreceptor for the photophobic response of B. japonicum. © 2006 Elsevier B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The primary photoprocesses in the photoreceptor for the step-up photophobic response of the light-adapted cells of Blepharisma japonicum (OBIP, OxyBlepharismin bInding Protein) have been studied by ultrafast UV-vis transient spectroscopy. The results are rationalized in terms of heterogeneity of the OBIP sample. Two independent classes of chromoprotein are proposed: a "reactive" species, which presents a specific 680-nm band decaying in 4 and 56 ps and a "non-reactive" one, which behaves like the free chromophore (OxyBP) in solution. A bimolecular photooxidation of OxyBP in the presence of 1,4-benzoquinone was performed to record the absorption spectrum of the OxyBP radical cation. Comparison with reactive OBIP suggests that an electron transfer could be involved in the primary photoprocesses of OBIP and possibly trigger the sensory transduction chain of B. japonicum. In addition, the specificity of the chromophore-protein interaction was investigated by studying the artificial complex that OxyBP forms with human serum albumin (HSA). OxyBP-HSA happens to be spectroscopically much closer to free OxyBP than to OBIP. This highlights the specific nature of the interaction between OxyBP and its native protein partner and further supports the proposal that OBIP is the actual photoreceptor for the photophobic response of B. japonicum. © 2006 Elsevier B.V. All rights reserved. |
Role of arginine 52 on the primary photoinduced events in the PYP photocycle Article de journal P Changenet-Barret; P Plaza; M M Martin; H Chosrowjan; S Taniguchi; N Mataga; Y Imamoto; M Kataoka Chemical Physics Letters, 434 (4-6), p. 320–325, 2007. @article{Changenet-Barret:2007,
title = {Role of arginine 52 on the primary photoinduced events in the PYP photocycle},
author = {P Changenet-Barret and P Plaza and M M Martin and H Chosrowjan and S Taniguchi and N Mataga and Y Imamoto and M Kataoka},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-33846203359&doi=10.1016%2fj.cplett.2006.12.027&partnerID=40&md5=63606ca56aff55ca2e30db6af88fa152},
doi = {10.1016/j.cplett.2006.12.027},
year = {2007},
date = {2007-01-01},
journal = {Chemical Physics Letters},
volume = {434},
number = {4-6},
pages = {320--325},
abstract = {We investigated the earliest steps of the photocycle of photoactive yellow protein (PYP) and its R52Q mutant in aqueous solution, by subpicosecond transient absorption spectroscopy. Our aim was to address the role of the positively charged Arg52 residue. We found that the relaxation mechanism of R52Q is similar to that of wt-PYP and discarded that Arg52 plays a key role in the chromophore photoreactivity. The excited-state decay of R52Q is however slower, confirming previous fluorescence up-conversion data. The experiments also reveal a slower stabilization of the cis isomer product. The loosening of the protein pocket and structural heterogeneities are discussed. © 2006 Elsevier B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We investigated the earliest steps of the photocycle of photoactive yellow protein (PYP) and its R52Q mutant in aqueous solution, by subpicosecond transient absorption spectroscopy. Our aim was to address the role of the positively charged Arg52 residue. We found that the relaxation mechanism of R52Q is similar to that of wt-PYP and discarded that Arg52 plays a key role in the chromophore photoreactivity. The excited-state decay of R52Q is however slower, confirming previous fluorescence up-conversion data. The experiments also reveal a slower stabilization of the cis isomer product. The loosening of the protein pocket and structural heterogeneities are discussed. © 2006 Elsevier B.V. All rights reserved. |
Target analysis of primary photoprocesses involved in the oxyblepharismin-binding protein Article de journal P Plaza; M Mahet; M M Martin; G Checcucci; F Lenci Journal of Physical Chemistry B, 111 (4), p. 690–696, 2007. @article{Plaza:2007,
title = {Target analysis of primary photoprocesses involved in the oxyblepharismin-binding protein},
author = {P Plaza and M Mahet and M M Martin and G Checcucci and F Lenci},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-33847050886&doi=10.1021%2fjp0642591&partnerID=40&md5=493aaa2ac98825d3cee9d4a879e14b4f},
doi = {10.1021/jp0642591},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physical Chemistry B},
volume = {111},
number = {4},
pages = {690--696},
abstract = {Target analysis is performed on previously published transient absorption spectra of the 200-kDa oxyblepharismin-binding protein (OBIP) thought to trigger the photophobic response of the ciliate Blepharisma japonicum. The OBIP sample is considered as heterogeneous and made of two distinct classes of chromophore-protein complexes. A so-called nonreactive class is seen to be comparable to free oxyblepharismin in organic solution. Another, reactive, class is shown to undergo a fast picosecond photocycle involving the formation in 4 ps of an intermediate state noted Y1. The spectrum associated to Y1 bears striking similarities with that of the oxyblepharismin radical cation. This element favors the hypothesis that an excited-state intermolecular electron-transfer could be the primary step of the sensory transduction chain of B. japonicum. Proton release is also considered as a possible secondary step. These possibilities support the idea that reactive OBIP functions like an electron or proton pump. We alternatively propose a new hypothesis stating that the fast photocycle of reactive OBIP actually does not generate any photoproduct or protein change of conformation but is involved in another biological function. It would act as a kind of solar screen, providing additional protection to the light-adapted form of B. japonicum in case of excessive illumination. © 2007 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Target analysis is performed on previously published transient absorption spectra of the 200-kDa oxyblepharismin-binding protein (OBIP) thought to trigger the photophobic response of the ciliate Blepharisma japonicum. The OBIP sample is considered as heterogeneous and made of two distinct classes of chromophore-protein complexes. A so-called nonreactive class is seen to be comparable to free oxyblepharismin in organic solution. Another, reactive, class is shown to undergo a fast picosecond photocycle involving the formation in 4 ps of an intermediate state noted Y1. The spectrum associated to Y1 bears striking similarities with that of the oxyblepharismin radical cation. This element favors the hypothesis that an excited-state intermolecular electron-transfer could be the primary step of the sensory transduction chain of B. japonicum. Proton release is also considered as a possible secondary step. These possibilities support the idea that reactive OBIP functions like an electron or proton pump. We alternatively propose a new hypothesis stating that the fast photocycle of reactive OBIP actually does not generate any photoproduct or protein change of conformation but is involved in another biological function. It would act as a kind of solar screen, providing additional protection to the light-adapted form of B. japonicum in case of excessive illumination. © 2007 American Chemical Society. |
Ultrafast light-induced response of photoactive yellow protein chromophore analogues Article de journal A Espagne; D H Paik; P Changenet-Barret; P Plaza; M M Martin; A H Zewail Photochemical and Photobiological Sciences, 6 (7), p. 780–787, 2007. @article{Espagne:2007,
title = {Ultrafast light-induced response of photoactive yellow protein chromophore analogues},
author = {A Espagne and D H Paik and P Changenet-Barret and P Plaza and M M Martin and A H Zewail},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-34447134184&doi=10.1039%2fb700927e&partnerID=40&md5=f30b6c18d177d6acc83123a44276d1be},
doi = {10.1039/b700927e},
year = {2007},
date = {2007-01-01},
journal = {Photochemical and Photobiological Sciences},
volume = {6},
number = {7},
pages = {780--787},
abstract = {The fluorescence decays of several analogues of the photoactive yellow protein (PYP) chromophore in aqueous solution have been measured by femtosecond fluorescence up-conversion and the corresponding time-resolved fluorescence spectra have been reconstructed. The native chromophore of PYP is a thioester derivative of p-coumaric acid in its trans deprotonated form. Fluorescence kinetics are reported for a thioester phenyl analogue and for two analogues where the thioester group has been changed to amide and carboxylate groups. The kinetics are compared to those we previously reported for the analogues bearing ketone and ester groups. The fluorescence decays of the full series are found to lie in the 1-10 ps range depending on the electron-acceptor character of the substituent, in good agreement with the excited-state relaxation kinetics extracted from transient absorption measurements. Steady-state photolysis is also examined and found to depend strongly on the nature of the substituent. While it has been shown that the ultrafast light-induced response of the chromophore in PYP is controlled by the properties of the protein nanospace, the present results demonstrate that, in solution, the relaxation dynamics and pathway of the chromophore is controlled by its electron donor-acceptor structure: structures of stronger electron donor-acceptor character lead to faster decays and less photoisomerisation. © The Royal Society of Chemistry and Owner Societies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The fluorescence decays of several analogues of the photoactive yellow protein (PYP) chromophore in aqueous solution have been measured by femtosecond fluorescence up-conversion and the corresponding time-resolved fluorescence spectra have been reconstructed. The native chromophore of PYP is a thioester derivative of p-coumaric acid in its trans deprotonated form. Fluorescence kinetics are reported for a thioester phenyl analogue and for two analogues where the thioester group has been changed to amide and carboxylate groups. The kinetics are compared to those we previously reported for the analogues bearing ketone and ester groups. The fluorescence decays of the full series are found to lie in the 1-10 ps range depending on the electron-acceptor character of the substituent, in good agreement with the excited-state relaxation kinetics extracted from transient absorption measurements. Steady-state photolysis is also examined and found to depend strongly on the nature of the substituent. While it has been shown that the ultrafast light-induced response of the chromophore in PYP is controlled by the properties of the protein nanospace, the present results demonstrate that, in solution, the relaxation dynamics and pathway of the chromophore is controlled by its electron donor-acceptor structure: structures of stronger electron donor-acceptor character lead to faster decays and less photoisomerisation. © The Royal Society of Chemistry and Owner Societies. |
Ultrafast Structural Dynamics of Water Induced by Dissipation of Vibrational Energy Article de journal S Ashihara; N Huse; A Espagne; E T J Nibbering; T Elsaesser Journal of Physical Chemistry A, 111 (5), p. 743-746, 2007, ISSN: 1089-5639. @article{RN53,
title = {Ultrafast Structural Dynamics of Water Induced by Dissipation of Vibrational Energy},
author = {S Ashihara and N Huse and A Espagne and E T J Nibbering and T Elsaesser},
doi = {10.1021/jp0676538},
issn = {1089-5639},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physical Chemistry A},
volume = {111},
number = {5},
pages = {743-746},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
2006
|
Excited-state dynamics of the PYP chromophore in solution. Environment and structure effects Inproceedings A Espagne; P Changenet-Barret; J -B Baudin; P Plaza; M M Martin Jr, Castleman A W; Kimble, M L (Ed.): VIIth International Conference on Femtochemistry, p. 204-214, Elsevier, 2006. @inproceedings{RN101,
title = {Excited-state dynamics of the PYP chromophore in solution. Environment and structure effects},
author = {A Espagne and P Changenet-Barret and J -B Baudin and P Plaza and M M Martin},
editor = {A W Castleman Jr and M L Kimble},
year = {2006},
date = {2006-01-01},
booktitle = {VIIth International Conference on Femtochemistry},
pages = {204-214},
publisher = {Elsevier},
series = {Femtochemistry VII: Fundamental Ultrafast Processes in Chemistry, Physics and Biology},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
|