2017
|
Repair of (6-4) Lesions in DNA by (6-4) Photolyase: 20 Years of Quest for the Photoreaction Mechanism Article de journal J Yamamoto; P Plaza; K Brettel Photochemistry and Photobiology, 93 (1), p. 51–66, 2017. @article{Yamamoto:2017a,
title = {Repair of (6-4) Lesions in DNA by (6-4) Photolyase: 20 Years of Quest for the Photoreaction Mechanism},
author = {J Yamamoto and P Plaza and K Brettel},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85011581630&doi=10.1111%2fphp.12696&partnerID=40&md5=4127461fd365fb839dfef3ed244ba606},
doi = {10.1111/php.12696},
year = {2017},
date = {2017-01-01},
journal = {Photochemistry and Photobiology},
volume = {93},
number = {1},
pages = {51--66},
abstract = {Exposure of DNA to ultraviolet (UV) light from the Sun or from other sources causes the formation of harmful and carcinogenic crosslinks between adjacent pyrimidine nucleobases, namely cyclobutane pyrimidine dimers and pyrimidine(6\textendash4)pyrimidone photoproducts. Nature has developed unique flavoenzymes, called DNA photolyases, that utilize blue light, that is photons of lower energy than those of the damaging light, to repair these lesions. In this review, we focus on the chemically challenging repair of the (6\textendash4) photoproducts by (6\textendash4) photolyase and describe the major events along the quest for the reaction mechanisms, over the 20 years since the discovery of (6-4) photolyase. © 2016 The American Society of Photobiology},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Exposure of DNA to ultraviolet (UV) light from the Sun or from other sources causes the formation of harmful and carcinogenic crosslinks between adjacent pyrimidine nucleobases, namely cyclobutane pyrimidine dimers and pyrimidine(6–4)pyrimidone photoproducts. Nature has developed unique flavoenzymes, called DNA photolyases, that utilize blue light, that is photons of lower energy than those of the damaging light, to repair these lesions. In this review, we focus on the chemically challenging repair of the (6–4) photoproducts by (6–4) photolyase and describe the major events along the quest for the reaction mechanisms, over the 20 years since the discovery of (6-4) photolyase. © 2016 The American Society of Photobiology |
Ultrafast flavin photoreduction in an oxidized animal (6-4) photolyase through an unconventional tryptophan tetrad Article de journal P R Martin; F Lacombat; A Espagne; N Dozova; P Plaza; J Yamamoto; P Müller; K Brettel; A De La Lande Physical Chemistry Chemical Physics, 19 (36), p. 24493–24504, 2017. @article{Martin:2017a,
title = {Ultrafast flavin photoreduction in an oxidized animal (6-4) photolyase through an unconventional tryptophan tetrad},
author = {P R Martin and F Lacombat and A Espagne and N Dozova and P Plaza and J Yamamoto and P M\"{u}ller and K Brettel and A De La Lande},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-85029817108&doi=10.1039%2fc7cp04555g&partnerID=40&md5=b5966e02f7c4f4fa9b385f15647311c3},
doi = {10.1039/c7cp04555g},
year = {2017},
date = {2017-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {19},
number = {36},
pages = {24493--24504},
abstract = {Photolyases are flavoenzymes repairing UV-induced lesions in DNA, which may be activated by a photoreduction of their FAD cofactor. In most photolyases, this photoreduction proceeds by electron transfer along a chain of three tryptophan (Trp) residues, connecting the flavin to the protein surface. Much less studied, animal (6-4) photolyases (repairing pyrimidine-pyrimidone (6-4) photoproducts) are particularly interesting as they were recently shown to have a longer electron transfer chain, counting four Trp residues. Using femtosecond polarized transient absorption spectroscopy, we performed a detailed analysis of the photoactivation reaction in the (6-4) photolyase of Xenopus laevis with oxidized FAD. We showed that the excited flavin is very quickly reduced (∼0.5 ps) by a nearby tryptophan residue, yielding FAD•- and WH•+ radicals. Subsequent kinetic steps in the picosecond regime were assigned to the migration of the positive charge along the Trp tetrad, in competition with charge recombination. We propose that the positive charge is actually delocalized over various Trp residues during most of the dynamics and that charge recombination essentially occurs through the proximal tryptophanyl radical. Oxidation of the fourth tryptophan is thought to be reached about as fast as that of the third one (∼40 ps), based on a comparison with a mutant protein lacking the distal Trp, implying ultrafast electron transfer between these two residues. This unusual mechanism sheds light on the rich diversity of electron transfer pathways found in various photolyases, and evolution-related cryptochromes alike. © the Owner Societies 2017.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Photolyases are flavoenzymes repairing UV-induced lesions in DNA, which may be activated by a photoreduction of their FAD cofactor. In most photolyases, this photoreduction proceeds by electron transfer along a chain of three tryptophan (Trp) residues, connecting the flavin to the protein surface. Much less studied, animal (6-4) photolyases (repairing pyrimidine-pyrimidone (6-4) photoproducts) are particularly interesting as they were recently shown to have a longer electron transfer chain, counting four Trp residues. Using femtosecond polarized transient absorption spectroscopy, we performed a detailed analysis of the photoactivation reaction in the (6-4) photolyase of Xenopus laevis with oxidized FAD. We showed that the excited flavin is very quickly reduced (∼0.5 ps) by a nearby tryptophan residue, yielding FAD•- and WH•+ radicals. Subsequent kinetic steps in the picosecond regime were assigned to the migration of the positive charge along the Trp tetrad, in competition with charge recombination. We propose that the positive charge is actually delocalized over various Trp residues during most of the dynamics and that charge recombination essentially occurs through the proximal tryptophanyl radical. Oxidation of the fourth tryptophan is thought to be reached about as fast as that of the third one (∼40 ps), based on a comparison with a mutant protein lacking the distal Trp, implying ultrafast electron transfer between these two residues. This unusual mechanism sheds light on the rich diversity of electron transfer pathways found in various photolyases, and evolution-related cryptochromes alike. © the Owner Societies 2017. |
2016
|
Excited-state symmetry breaking of linear quadrupolar chromophores: A transient absorption study Article de journal N Dozova; L Ventelon; G Clermont; M Blanchard-Desce; P Plaza Chemical Physics Letters, 664 , p. 56–62, 2016. @article{Dozova:2016,
title = {Excited-state symmetry breaking of linear quadrupolar chromophores: A transient absorption study},
author = {N Dozova and L Ventelon and G Clermont and M Blanchard-Desce and P Plaza},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84991648542&doi=10.1016%2fj.cplett.2016.10.020&partnerID=40&md5=93f48fbad7f0a0a40b036bef2e73147a},
doi = {10.1016/j.cplett.2016.10.020},
year = {2016},
date = {2016-01-01},
journal = {Chemical Physics Letters},
volume = {664},
pages = {56--62},
abstract = {The photophysical properties of two highly symmetrical quadrupolar chromophores were studied by both steady-state and transient absorption spectroscopy. Their excited-state behavior is dominated by the solvent-induced Stokes shift of the stimulated-emission band. The origin of this shift is attributed to symmetry breaking that confers a non-vanishing dipole moment to the excited state of both compounds. This dipole moment is large and constant in DMSO, whereas symmetry breaking appears significantly slower and leading to smaller excited-state dipole in toluene. Time-dependant increase of the excited-state dipole moment induced by weak solvation is proposed to explain the results in toluene. © 2016 Elsevier B.V.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The photophysical properties of two highly symmetrical quadrupolar chromophores were studied by both steady-state and transient absorption spectroscopy. Their excited-state behavior is dominated by the solvent-induced Stokes shift of the stimulated-emission band. The origin of this shift is attributed to symmetry breaking that confers a non-vanishing dipole moment to the excited state of both compounds. This dipole moment is large and constant in DMSO, whereas symmetry breaking appears significantly slower and leading to smaller excited-state dipole in toluene. Time-dependant increase of the excited-state dipole moment induced by weak solvation is proposed to explain the results in toluene. © 2016 Elsevier B.V. |
Ultrafast Dynamics of a Green Fluorescent Protein Chromophore Analogue: Competition between Excited-State Proton Transfer and Torsional Relaxation Article de journal T Chatterjee; F Lacombat; D Yadav; M Mandal; P Plaza; A Espagne; P K Mandal Journal of Physical Chemistry B, 120 (36), p. 9716–9722, 2016. @article{Chatterjee:2016,
title = {Ultrafast Dynamics of a Green Fluorescent Protein Chromophore Analogue: Competition between Excited-State Proton Transfer and Torsional Relaxation},
author = {T Chatterjee and F Lacombat and D Yadav and M Mandal and P Plaza and A Espagne and P K Mandal},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84987847663&doi=10.1021%2facs.jpcb.6b05795&partnerID=40&md5=dbc7a71ee9d3e1a62872f19c1f0c7204},
doi = {10.1021/acs.jpcb.6b05795},
year = {2016},
date = {2016-01-01},
journal = {Journal of Physical Chemistry B},
volume = {120},
number = {36},
pages = {9716--9722},
abstract = {The competition between excited-state proton transfer (ESPT) and torsion plays a central role in the photophysics of fluorescent proteins of the green fluorescent protein (GFP) family and their chromophores. Here, it was investigated in a single GFP chromophore analogue bearing o-hydroxy and p-diethylamino substituents, OHIM. The light-induced dynamics of OHIM was studied by femtosecond transient absorption spectroscopy, at different pH. We found that the photophysics of OHIM is determined by the electron-donating character of the diethylamino group: torsional relaxation dominates when the diethylamino group is neutral, whereas ultrafast ESPT followed by cis/trans isomerization and ground-state reprotonation are observed when the diethylamino group is protonated and therefore inactive as an electron donor. © 2016 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The competition between excited-state proton transfer (ESPT) and torsion plays a central role in the photophysics of fluorescent proteins of the green fluorescent protein (GFP) family and their chromophores. Here, it was investigated in a single GFP chromophore analogue bearing o-hydroxy and p-diethylamino substituents, OHIM. The light-induced dynamics of OHIM was studied by femtosecond transient absorption spectroscopy, at different pH. We found that the photophysics of OHIM is determined by the electron-donating character of the diethylamino group: torsional relaxation dominates when the diethylamino group is neutral, whereas ultrafast ESPT followed by cis/trans isomerization and ground-state reprotonation are observed when the diethylamino group is protonated and therefore inactive as an electron donor. © 2016 American Chemical Society. |
2015
|
Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases Article de journal P Müller; J Yamamoto; P R Martin; S Iwai; K Brettel Chemical Communications, 51 (85), p. 15502-15505, 2015, ISSN: 1359-7345. @article{RN116,
title = {Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases},
author = {P M\"{u}ller and J Yamamoto and P R Martin and S Iwai and K Brettel},
url = {<Go to ISI>://WOS:000363167900005},
doi = {10.1039/c5cc06276d},
issn = {1359-7345},
year = {2015},
date = {2015-01-01},
journal = {Chemical Communications},
volume = {51},
number = {85},
pages = {15502-15505},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
Photo-induced cation translocation in a molecular shuttle based on a calix[4]-biscrown including DCM and DMABN chromophores Article de journal N Dozova; R Kumar; T Pradhan; F Lacombat; B Valeur; J S Kim; P Plaza Chemical Communications, 51 (80), p. 14859–14861, 2015. @article{Dozova:2015,
title = {Photo-induced cation translocation in a molecular shuttle based on a calix[4]-biscrown including DCM and DMABN chromophores},
author = {N Dozova and R Kumar and T Pradhan and F Lacombat and B Valeur and J S Kim and P Plaza},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84942474410&doi=10.1039%2fc5cc06054k&partnerID=40&md5=857b5558380ed42e21e82790184d89cb},
doi = {10.1039/c5cc06054k},
year = {2015},
date = {2015-01-01},
journal = {Chemical Communications},
volume = {51},
number = {80},
pages = {14859--14861},
abstract = {We present a new molecular shuttle, consisting of a calixarene core attached to two different photoactive centers, DCM and DMABN. We show that a K+ ion bound to the DCM-grafted crown is translocated towards the other site of the molecule upon photoexcitation, but not released to the bulk. © The Royal Society of Chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We present a new molecular shuttle, consisting of a calixarene core attached to two different photoactive centers, DCM and DMABN. We show that a K+ ion bound to the DCM-grafted crown is translocated towards the other site of the molecule upon photoexcitation, but not released to the bulk. © The Royal Society of Chemistry. |
Photoswitching Kinetics and Phase-Sensitive Detection Add Discriminative Dimensions for Selective Fluorescence Imaging Article de journal Jérôme Querard; Tal-Zvi Markus; Marie-Aude Plamont; Carole Gauron; Pengcheng Wang; Agathe Espagne; Michel Volovitch; Sophie Vriz; Vincent Croquette; Arnaud Gautier; Thomas Le Saux; Ludovic Jullien Angewandte Chemie International Edition, 54 (9), p. 2633-2637, 2015, ISSN: 1433-7851. @article{RN43b,
title = {Photoswitching Kinetics and Phase-Sensitive Detection Add Discriminative Dimensions for Selective Fluorescence Imaging},
author = {J\'{e}r\^{o}me Querard and Tal-Zvi Markus and Marie-Aude Plamont and Carole Gauron and Pengcheng Wang and Agathe Espagne and Michel Volovitch and Sophie Vriz and Vincent Croquette and Arnaud Gautier and Thomas Le Saux and Ludovic Jullien},
doi = {10.1002/anie.201408985},
issn = {1433-7851},
year = {2015},
date = {2015-01-01},
journal = {Angewandte Chemie International Edition},
volume = {54},
number = {9},
pages = {2633-2637},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|
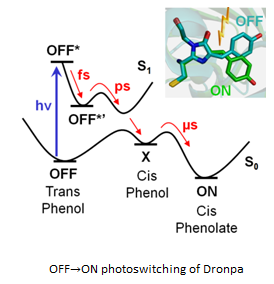
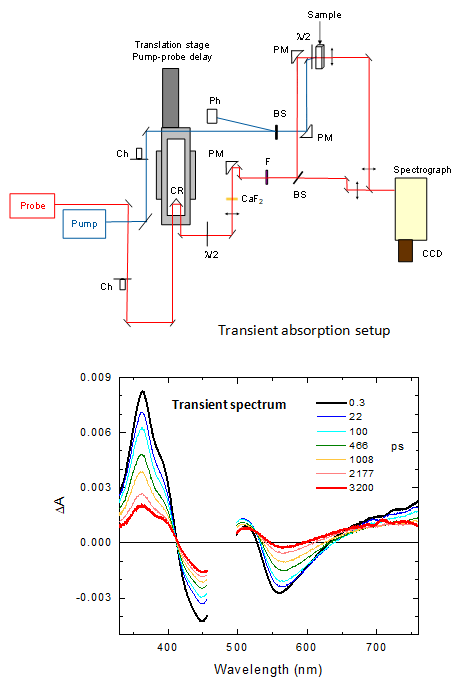
Real-time monitoring of chromophore isomerization and deprotonation during the photoactivation of the fluorescent protein Dronpa Article de journal D Yadav; F Lacombat; N Dozova; F Rappaport; P Plaza; A Espagne Journal of Physical Chemistry B, 119 (6), p. 2404–2414, 2015. @article{Yadav:2015,
title = {Real-time monitoring of chromophore isomerization and deprotonation during the photoactivation of the fluorescent protein Dronpa},
author = {D Yadav and F Lacombat and N Dozova and F Rappaport and P Plaza and A Espagne},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84922794115&doi=10.1021%2fjp507094f&partnerID=40&md5=6618718bf1ba6e1f5e5f2194c9480d5b},
doi = {10.1021/jp507094f},
year = {2015},
date = {2015-01-01},
journal = {Journal of Physical Chemistry B},
volume = {119},
number = {6},
pages = {2404--2414},
abstract = {Dronpa is a photochromic green fluorescent protein (GFP) homologue used as a probe in super-resolution microscopy. It is known that the photochromic reaction involves cis/trans isomerization of the chromophore and protonation/deprotonation of its phenol group, but the sequence in time of the two steps and their characteristic time scales are still the subject of much debate. We report here a comprehensive UV-visible transient absorption spectroscopy study of the photoactivation mechanism of Dronpa, covering all relevant time scales from ∼100 fs to milliseconds. The Dronpa-2 variant was also studied and showed the same behavior. By carefully controlling the excitation energy to avoid multiphoton processes, we could measure both the spectrum and the anisotropy of the first photoactivation intermediate. We show that the observed few nanometer blue-shift of this intermediate is characteristic for a neutral cis chromophore, and that its anisotropy of ∼0.2 is in good agreement with the reorientation of the transition dipole moment expected upon isomerization. These data constitute the first clear evidence that trans → cis isomerization of the chromophore precedes its deprotonation and occurs on the picosecond time scale, concomitantly to the excited-state decay. We found the deprotonation step to follow in ∼10 μs and lead directly from the neutral cis intermediate to the final state. © 2014 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dronpa is a photochromic green fluorescent protein (GFP) homologue used as a probe in super-resolution microscopy. It is known that the photochromic reaction involves cis/trans isomerization of the chromophore and protonation/deprotonation of its phenol group, but the sequence in time of the two steps and their characteristic time scales are still the subject of much debate. We report here a comprehensive UV-visible transient absorption spectroscopy study of the photoactivation mechanism of Dronpa, covering all relevant time scales from ∼100 fs to milliseconds. The Dronpa-2 variant was also studied and showed the same behavior. By carefully controlling the excitation energy to avoid multiphoton processes, we could measure both the spectrum and the anisotropy of the first photoactivation intermediate. We show that the observed few nanometer blue-shift of this intermediate is characteristic for a neutral cis chromophore, and that its anisotropy of ∼0.2 is in good agreement with the reorientation of the transition dipole moment expected upon isomerization. These data constitute the first clear evidence that trans → cis isomerization of the chromophore precedes its deprotonation and occurs on the picosecond time scale, concomitantly to the excited-state decay. We found the deprotonation step to follow in ∼10 μs and lead directly from the neutral cis intermediate to the final state. © 2014 American Chemical Society. |
2013
|
Repair of the (6-4) photoproduct by DNA photolyase requires two photons Article de journal J Yamamoto; P R Martin; S Iwai; P Plaza; K Brettel Angewandte Chemie - International Edition, 52 (29), p. 7432–7436, 2013. @article{Yamamoto:2013,
title = {Repair of the (6-4) photoproduct by DNA photolyase requires two photons},
author = {J Yamamoto and P R Martin and S Iwai and P Plaza and K Brettel},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84880097261&doi=10.1002%2fanie.201301567&partnerID=40&md5=e6cc2dbe1d4d7ec08c3ac0336fbecaf2},
doi = {10.1002/anie.201301567},
year = {2013},
date = {2013-01-01},
journal = {Angewandte Chemie - International Edition},
volume = {52},
number = {29},
pages = {7432--7436},
abstract = {It takes two (photons) to tango: Single-turnover flash experiments showed that the flavoenzyme (6-4) photolyase uses a successive two-photon mechanism to repair the UV-induced T(6-4)T lesion in DNA (see picture). The intermediate (X) formed by the first photoreaction is likely to be the oxetane-bridged dimer T(ox)T. The enzyme could stabilize the normally short-lived T(ox)T, allowing repair to be completed by the second photoreaction. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
It takes two (photons) to tango: Single-turnover flash experiments showed that the flavoenzyme (6-4) photolyase uses a successive two-photon mechanism to repair the UV-induced T(6-4)T lesion in DNA (see picture). The intermediate (X) formed by the first photoreaction is likely to be the oxetane-bridged dimer T(ox)T. The enzyme could stabilize the normally short-lived T(ox)T, allowing repair to be completed by the second photoreaction. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. |
Ultrafast carbonyl motion of the photoactive yellow protein chromophore probed by femtosecond circular dichroism Article de journal L Mendonça; F Hache; P Changenet-Barret; P Plaza; H Chosrowjan; S Taniguchi; Y Imamoto Journal of the American Chemical Society, 135 (39), p. 14637–14643, 2013. @article{Mendonca:2013,
title = {Ultrafast carbonyl motion of the photoactive yellow protein chromophore probed by femtosecond circular dichroism},
author = {L Mendon\c{c}a and F Hache and P Changenet-Barret and P Plaza and H Chosrowjan and S Taniguchi and Y Imamoto},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84885113374&doi=10.1021%2fja404503q&partnerID=40&md5=b58dd62402f3e9bde273577c40cea045},
doi = {10.1021/ja404503q},
year = {2013},
date = {2013-01-01},
journal = {Journal of the American Chemical Society},
volume = {135},
number = {39},
pages = {14637--14643},
abstract = {Motions of the trans-p-coumaric acid carbonyl group following the photoexcitation of the R52Q mutant of photoactive yellow protein (PYP) are investigated, for the first time, by ultrafast time-resolved circular dichroism (TRCD) spectroscopy. TRCD is monitored in the near-ultraviolet, over a time scale of 10 ps. Immediately after excitation, TRCD is found to exhibit a large negative peak, which decays within a few picoseconds. A quantitative analysis of the signals shows that, upon excitation, the carbonyl group undergoes a fast (\^{a}‰o0.8 ps) and unidirectional flipping motion in the excited state with an angle of ca. 17-53. For the subset of proteins that do not enter the signaling photocycle, TRCD provides strong evidence that the carbonyl group moves back to its initial position, leading to the formation of a nonreactive ground-state intermediate of trans conformation. The initial ground state is then restored within ca. 3 ps. Comparative study of R52Q and wild-type PYP provides direct evidence that the absence of Arg52 has no effect on the conformational changes of the chromophore during those steps. © 2013 American Chemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Motions of the trans-p-coumaric acid carbonyl group following the photoexcitation of the R52Q mutant of photoactive yellow protein (PYP) are investigated, for the first time, by ultrafast time-resolved circular dichroism (TRCD) spectroscopy. TRCD is monitored in the near-ultraviolet, over a time scale of 10 ps. Immediately after excitation, TRCD is found to exhibit a large negative peak, which decays within a few picoseconds. A quantitative analysis of the signals shows that, upon excitation, the carbonyl group undergoes a fast (â‰o0.8 ps) and unidirectional flipping motion in the excited state with an angle of ca. 17-53. For the subset of proteins that do not enter the signaling photocycle, TRCD provides strong evidence that the carbonyl group moves back to its initial position, leading to the formation of a nonreactive ground-state intermediate of trans conformation. The initial ground state is then restored within ca. 3 ps. Comparative study of R52Q and wild-type PYP provides direct evidence that the absence of Arg52 has no effect on the conformational changes of the chromophore during those steps. © 2013 American Chemical Society. |
2012
|
Method For Generating Cyan Fluorescent Proteins Which Have A Reduced Ph Sensitivity Patent Pasquier Helene; Merola Fabienne; Erard Marie; Espagne Agathe; Fredj Asma WO 2012/172095 A1, 2012. @patent{lens.org/080-732-449-078-090,
title = {Method For Generating Cyan Fluorescent Proteins Which Have A Reduced Ph Sensitivity},
author = {Pasquier Helene and Merola Fabienne and Erard Marie and Espagne Agathe and Fredj Asma},
url = {https://lens.org/080-732-449-078-090},
year = {2012},
date = {2012-12-20},
number = {WO 2012/172095 A1},
location = {World Intellectual Property Organization},
keywords = {},
pubstate = {published},
tppubtype = {patent}
}
|
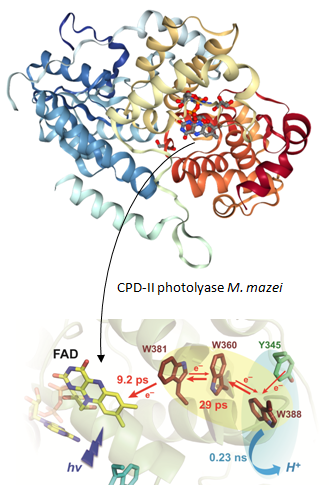
Photoantenna in two cryptochrome-photolyase proteins from O. tauri: Presence, nature and ultrafast photoinduced dynamics Article de journal J Brazard; C Ley; F Lacombat; P Plaza; L Mony; M Heijde; G Zabulon; C Bowler Journal of Photochemistry and Photobiology A: Chemistry, 234 , p. 135–145, 2012. @article{Brazard:2012,
title = {Photoantenna in two cryptochrome-photolyase proteins from O. tauri: Presence, nature and ultrafast photoinduced dynamics},
author = {J Brazard and C Ley and F Lacombat and P Plaza and L Mony and M Heijde and G Zabulon and C Bowler},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84859544483&doi=10.1016%2fj.jphotochem.2012.01.012&partnerID=40&md5=99a665d61b945b52ae6748e042d2b835},
doi = {10.1016/j.jphotochem.2012.01.012},
year = {2012},
date = {2012-01-01},
journal = {Journal of Photochemistry and Photobiology A: Chemistry},
volume = {234},
pages = {135--145},
abstract = {Cryptochromes and photolyases are homologous flavoproteins either involved in photosensory functions or in the photorepair of UV-damaged DNA. We report on the presence, nature and ultrafast photoinduced dynamics of a photoantenna in two recently discovered cryptochrome/photolyase proteins (OtCPF1, a (6-4) photolyase, and OtCPF2, a cryptochrome-DASH), coming from the green alga Ostreococcus tauri. Whereas OtCPF1 does not show any apparent photoantenna after purification, OtCPF2 is found to bind the folate MTHF. Resonance energy transfer, from MTHF to the flavin cofactor (FAD) of OtCPF2, was studied by femtosecond transient absorption spectroscopy. It takes place in 15 ps when FAD is oxidized and 100 ps when it is fully reduced. The intrinsic photophysics of MTHF in acidic solution was studied for comparison. Two decays in the sub-100 ps regime were tentatively attributed to the presence of two conformers or to a quenching mechanism involving large-amplitude motions of the 1,2,3,4-tetrahydropyraxine ring. On the other hand, the nature of the missing photoantenna of OtCFP1 was questioned by means of sequence alignment and homology modeling. We propose that the deazaflavin 8-HDF could be the photoantenna of OtCFP1 in vivo. © 2012 Elsevier B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cryptochromes and photolyases are homologous flavoproteins either involved in photosensory functions or in the photorepair of UV-damaged DNA. We report on the presence, nature and ultrafast photoinduced dynamics of a photoantenna in two recently discovered cryptochrome/photolyase proteins (OtCPF1, a (6-4) photolyase, and OtCPF2, a cryptochrome-DASH), coming from the green alga Ostreococcus tauri. Whereas OtCPF1 does not show any apparent photoantenna after purification, OtCPF2 is found to bind the folate MTHF. Resonance energy transfer, from MTHF to the flavin cofactor (FAD) of OtCPF2, was studied by femtosecond transient absorption spectroscopy. It takes place in 15 ps when FAD is oxidized and 100 ps when it is fully reduced. The intrinsic photophysics of MTHF in acidic solution was studied for comparison. Two decays in the sub-100 ps regime were tentatively attributed to the presence of two conformers or to a quenching mechanism involving large-amplitude motions of the 1,2,3,4-tetrahydropyraxine ring. On the other hand, the nature of the missing photoantenna of OtCFP1 was questioned by means of sequence alignment and homology modeling. We propose that the deazaflavin 8-HDF could be the photoantenna of OtCFP1 in vivo. © 2012 Elsevier B.V. All rights reserved. |
Photophysics and Photochemistry of Supramolecular Systems incollection B Valeur; M -N Berberan-Santos; M M Martin; P Plaza Analytical Methods in Supramolecular Chemistry, Volume 1 & 2: Second Edition, 1 , p. 287–336, 2012. @incollection{Valeur:2012,
title = {Photophysics and Photochemistry of Supramolecular Systems},
author = {B Valeur and M -N Berberan-Santos and M M Martin and P Plaza},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84882348246&doi=10.1002%2f9783527644131.ch7&partnerID=40&md5=828ac5cbce13769dc5a7647bb02646a9},
doi = {10.1002/9783527644131.ch7},
year = {2012},
date = {2012-01-01},
booktitle = {Analytical Methods in Supramolecular Chemistry, Volume 1 & 2: Second Edition},
volume = {1},
pages = {287--336},
keywords = {},
pubstate = {published},
tppubtype = {incollection}
}
|
Reaction-coordinate tracking in the excited-state deactivation of the photoactive yellow protein chromophore in solution Article de journal P Changenet-Barret; F Lacombat; P Plaza Journal of Photochemistry and Photobiology A: Chemistry, 234 , p. 171–180, 2012. @article{Changenet-Barret:2012,
title = {Reaction-coordinate tracking in the excited-state deactivation of the photoactive yellow protein chromophore in solution},
author = {P Changenet-Barret and F Lacombat and P Plaza},
url = {https://www.scopus.com/inward/record.uri?eid=2-s2.0-84859535666&doi=10.1016%2fj.jphotochem.2012.03.011&partnerID=40&md5=79ecc65ea3cda63c045f288c70d56329},
doi = {10.1016/j.jphotochem.2012.03.011},
year = {2012},
date = {2012-01-01},
journal = {Journal of Photochemistry and Photobiology A: Chemistry},
volume = {234},
pages = {171--180},
abstract = {The earliest steps of the photoactive yellow protein (PYP) photocycle are known to involve cis-trans photoisomerization of its chromophore, the deprotonated trans-p-coumaric acid. In aqueous solution PYP chromophore analogues bearing the same thioester function as in the protein however do not isomerize and restore the initial trans configuration via a short-lived charge-transfer intermediate. In order to gain further insight into the nature of this non-radiative process, we report the first study by femtosecond transient absorption spectroscopy of a ketone derivative of the trans-p-coumaric acid, pCK -, which has been used as a PYP chromophore model in recent theoretical studies. While the transient spectra of pCK - in basic aqueous solution are similar to those of the thioester derivatives, we identify in decanol an additional deactivation route. It involves the formation of photoproduct with a lifetime of a few seconds, which we attribute to the cis isomer. These results are discussed in terms of both static and dynamic solvent effects on the excited-state deactivation. © 2012 Elsevier B.V. All rights reserved.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The earliest steps of the photoactive yellow protein (PYP) photocycle are known to involve cis-trans photoisomerization of its chromophore, the deprotonated trans-p-coumaric acid. In aqueous solution PYP chromophore analogues bearing the same thioester function as in the protein however do not isomerize and restore the initial trans configuration via a short-lived charge-transfer intermediate. In order to gain further insight into the nature of this non-radiative process, we report the first study by femtosecond transient absorption spectroscopy of a ketone derivative of the trans-p-coumaric acid, pCK -, which has been used as a PYP chromophore model in recent theoretical studies. While the transient spectra of pCK - in basic aqueous solution are similar to those of the thioester derivatives, we identify in decanol an additional deactivation route. It involves the formation of photoproduct with a lifetime of a few seconds, which we attribute to the cis isomer. These results are discussed in terms of both static and dynamic solvent effects on the excited-state deactivation. © 2012 Elsevier B.V. All rights reserved. |
2011
|
Cyan Fluorescent Protein Carries a Constitutive Mutation That Prevents Its Dimerization Article de journal A Espagne; M Erard; K Madiona; V Derrien; G Jonasson; B Levy; H Pasquier; R Melki; F Merola Biochemistry, 50 (4), p. 437-439, 2011, ISSN: 0006-2960. @article{RN29b,
title = {Cyan Fluorescent Protein Carries a Constitutive Mutation That Prevents Its Dimerization},
author = {A Espagne and M Erard and K Madiona and V Derrien and G Jonasson and B Levy and H Pasquier and R Melki and F Merola},
doi = {10.1021/bi1015875},
issn = {0006-2960},
year = {2011},
date = {2011-01-01},
journal = {Biochemistry},
volume = {50},
number = {4},
pages = {437-439},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
|