





Actualités
Voir toutes les actualités
2026 Summer Chemistry Day
Save the date ! Summer Chemistry Day is returning on Monday, June 29th 2026 !

Gerard Ferey: Materials from Institut Lavoisier Legacy Workshop

Signal strapping—a proteomic search technique for the discovery of metalloproteins

Journée Bram 2026 - 16 janvier
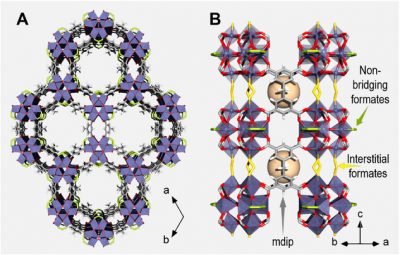
New article on photoaccumulation of long-lived electrons in a Ti-MOF

Winter Chemistry Day (for CPCV and IMAP members)
This year's Winter Chemistry Day (reserved to CPCV, IMAP and Chemistry Department members) is held on December 15th and for once, you will not get only chemistry presentations!
Recherche et formation
 |
Suivez-nous
| Tweets by Chimie__ENS |