





Actualités
Voir toutes les actualités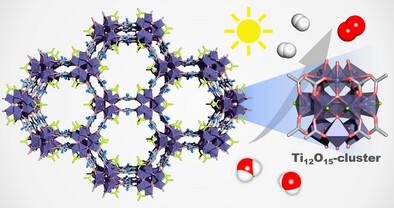
A Novel Ti12-Based Metal-Organic Framework for Photocatalytic Hydrogen Evolution

Bio-inspired Electrocatalysts for Water Oxidation

From Automated High-Throughput Experimentation to Self-Driving Laboratories: The Swiss Cat⁺ Experience

Strategies for Selective Chemical Sensing and Imaging with Polymeric Nanospheres

A Quest for Bio-Orthogonal Transition Metal Catalysis in Living Cells
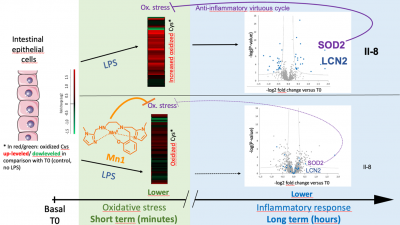
Kinetic Redox Shotgun Proteomics Reveals Specific Lipopolysaccharide Effects on Intestinal Epithelial Cells, Mitigated by a Mn Superoxide Dismutase Mimic
Recherche et formation
 |
 |
 |
 |
Suivez-nous
| Tweets by Chimie__ENS |