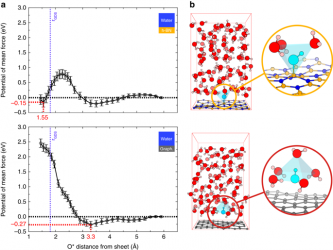
The recent emergence of nanofluidics has highlighted the exceptional properties of graphene and its boron-nitride counterpart as confining nanomaterials for water and ion transport. Surprisingly, ionic transport experiments have unveiled a consequent electrification of the water/carbon surfaces, with a contrasting response for its water/boron-nitride homologue. In this paper, we report free energy calculations based on ab initio molecular dynamics simulations of hydroxide OH− ions in water near graphene and hexagonal boron nitride (h-BN) layers. Our results disclose that both surfaces get charged through hydroxide adsorption, but two strongly different mechanisms are evidenced. The hydroxide species shows weak physisorption on the graphene surface while it exhibits also strong chemisorption on the h-BN surface. Interestingly OH− is shown to keep very fast lateral dynamics and interfacial mobility within the physisorbed layer on graphene. Taking into account the large ionic surface conductivity, an analytic transport model allows to reproduce quantitatively the experimental data.

N’hésitez pas à consulter le communiqué de presse associé à cet article : Graphène et Bore-Azote hexagonal : des faux-jumeaux innovants pour produire l’énergie bleue !
References:
Versatile Electrification of Two-dimensional Nanomaterials in Water
Benoît Grosjean, Marie-Laure Bocquet, and Rodolphe Vuilleumier
Nature Communications 10,1656 (2019)
DOI: 10.1038/s41467-019-09708-7
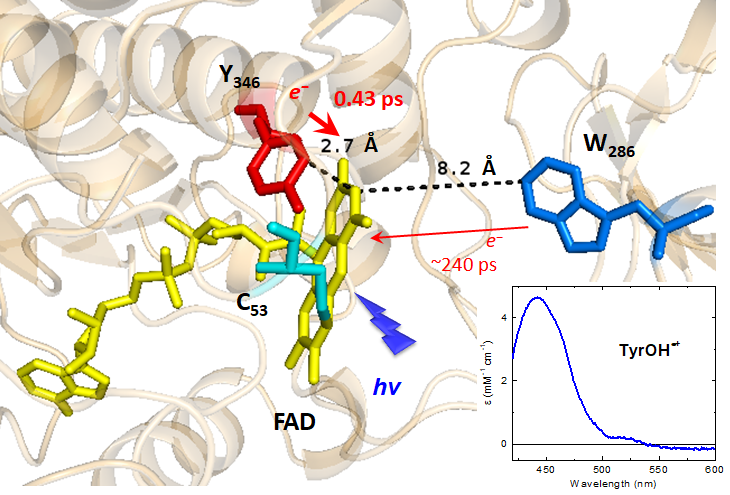
Flavoproteins often stabilize their flavin coenzyme by stacking interactions involving the isoalloxazine moiety of the flavin and an aromatic residue from the apoprotein. The bacterial FAD and folate-dependent tRNA methyltransferase TrmFO has the unique property of stabilizing its FAD coenzyme by an unusual H-bond-assisted π–π stacking interaction, involving a conserved tyrosine (Y346 in Bacillus subtilis TrmFO, BsTrmFO), the isoalloxazine of FAD and the backbone of a catalytic cysteine (C53). Here, the interaction between FAD and Y346has been investigated by measuring the photoinduced flavin dynamics of BsTrmFO in the wild-type (WT) protein, C53A and several Y346 mutants by ultrafast transient absorption spectroscopy. In C53A, the excited FAD very rapidly (0.43 ps) abstracts an electron from Y346, yielding the FAD˙−/Y346OH˙+ radical pair, while relaxation of the local environment (1.3 ps) of the excited flavin produces a slight Stokes shift of its stimulated emission band. The radical pair then decays via charge recombination, mostly in 3–4 ps, without any deprotonation of the Y346OH˙+ radical. Presumably, the H-bond between Y346 and the amide group of C53 increases the pKa of Y346OH˙+ and slows down its deprotonation. The dynamics of WT BsTrmFO shows additional slow decay components (43 and 700 ps), absent in the C53A mutant, assigned to excited FADox populations not undergoing fast photoreduction. Their presence is likely due to a more flexible structure of the WT protein, favored by the presence of C53. Interestingly, mutations of Y346 canceling its electron donating character lead to multiple slower quenching channels in the ps–ns regime. These channels are proposed to be due to electron abstraction either (i) from the adenine moiety of FAD, a distribution of the isoalloxazine–adenine distance in the absence of Y346 explaining the multiexponential decay, or (ii) from the W286 residue, possibly accounting for one of the decays. This work supports the idea that H-bond-assisted π–π stacking controls TrmFO’s active site dynamics, required for competent orientation of the reactive centers during catalysis.

Reference:
Ultrafast photoinduced flavin dynamics in the unusual active site of the tRNA methyltransferase TrmFO
Nadia Dozova, Fabien Lacombat, Charles Bou-Nader, Djemel Hamdane* and Pascal Plaza*
Phys. Chem. Chem. Phys., 2019, 21, 8743-8756
DOI: 10.1039/C8CP06072J
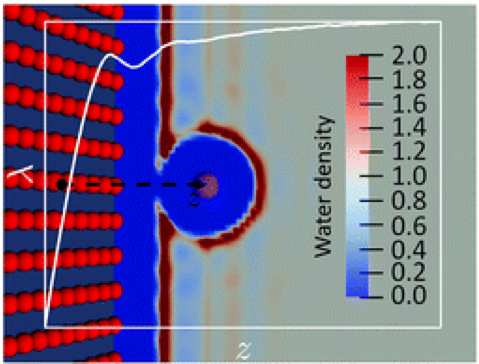
Beyond the dielectric continuum description initiated by Marcus theory, the standard theoretical approach to study electron transfer (ET) reactions in solution or at interfaces is to use classical force field or ab initio molecular dynamics simulations. We present here an alternative method based on liquid-state theory, namely molecular density functional theory, which is numerically much more efficient than simulations while still retaining the molecular nature of the solvent. We begin by reformulating molecular ET theory in a density functional language and show how to compute the various observables characterizing ET reactions from an ensemble of density functional minimizations. In particular, we define within that formulation the relevant order parameter of the reaction, the so-called vertical energy gap, and determine the Marcus free energy curves of both reactant and product states along that coordinate. Important thermodynamic quantities such as the reaction free energy and the reorganization free energies follow. We assess the validity of the method by studying the model Cl0 / Cl+ and Cl0 / Cl- ET reactions in bulk water for which molecular dynamics results are available. The anionic case is found to violate the standard Marcus theory. Finally, we take advantage of the computational efficiency of the method to study the influence of a solid–solvent interface on the ET, by investigating the evolution of the reorganization free energy of the Cl0 / Cl+ reaction when the atom approaches an atomistically resolved wall.

N’hésitez pas à consulter le communiqué de presse associé à cet article : Quand la théorie de Marcus se confronte à la DFT !
References :
A molecular density functional theory approach to electron transfer reactions
Guillaume Jeanmairet, Benjamin Rotenberg, Maximilien Levesque, Daniel Borgis and Mathieu Salanne
Chem. Sci., 2019, Advance Article
DOI: 10.1039/C8SC04512G
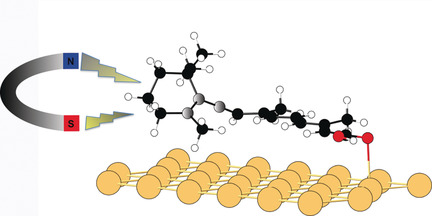
Inert metal surfaces present more chances of hosting organic intact radicals than other substrates, but large amounts of delocalized electronic states favor charge transfer and thus spin quenching. Lowering the molecule–substrate interaction is a usual strategy to stabilize radicals on surfaces. In some works, thin insulating layers were introduced to provide a controllable degree of electronic decoupling. Recently, retinoid molecules adsorbed on gold have been manipulated with a scanning tunneling microscope (STM) to exhibit a localized spin, but calculations failed to find a radical derivative of the molecule on the surface. Now the formation of a neutral radical spatially localized in a tilted and lifted cyclic end of the molecule is presented. An allene moiety provokes a perpendicular tilt of the cyclic end relative to the rest of the conjugated chain, thus localizing the spin of the dehydrogenated allene in its lifted subpart. DFT calculations and STM manipulations give support to the proposed mechanism.

N’hésitez pas à consulter le communiqué de presse associé à cet article :
Quand des molécules non-magnétiques organiques se transforment en aimant !
Également un communiqué de presse (en anglais) provenant de Mapping Ignorance :
Spin in a closed-shell organic molecule stabilized on a metallic surface
References:
Spin in a Closed-Shell Organic Molecule on a Metal Substrate Generated by a Sigmatropic Reaction
Marie-Laure Bocquet, Nicolas Lorente, Richard Berndt, and Manuel Gruber
Angew. Chem. Int. Ed. 2019, 58, 821
DOI: 10.1002/anie.201812121

Damien Baigl est lauréat du Prix Mergier-Bourdeix 2018 décerné par l’Académie des Sciences «alternativement dans le ressort de la division des sciences mathématiques et physiques, sciences de l’univers et leurs applications, et dans celui de la division des sciences chimiques, biologiques, médicales et leurs applications, à un.e jeune chercheur.e français.e se consacrant à des recherches fondamentales n’ayant aucun but lucratif, ne visant pas d’application immédiate et dont les résultats révèlent des dons exceptionnels».


Dr. Alexander Oleinick is the first recipient of Wuthrich International Young Star Award. The prize is named after the 2002 Nobel Chemistry Laureate Kurt Wuthrich and is awarded to « a young scientist for outstanding scientific performance during the early career stages« .

Alexander Oleinick with Kurt Wuthrich during the ceremony !

We demonstrate that molecular vibrations with originally low or zero intensity in a vibrational circular dichroism (VCD) spectrum attain chirality in molecular crystals by coordinated motion of the atoms. Ab initio molecular dynamics simulations of anharmonic solid-state VCD spectra of l-alanine crystals reveal how coherent vibrational modes exploit the space groupQs chirality, leading to nonlocal, enhanced VCD features, most significantly in the carbonyl region of the spectrum. The VCD-enhanced signal is ascribed to a helical arrangement of the oscillators in the crystal layers. No structural irregularities need to be considered to explain the amplification, but a crucial point lies in the polarization of charge, which requires an accurate description of the electronic structure. Delivering a quantitative atomic conception of supramolecular chirality induction, our ab initio scheme is applicable well beyond molecular crystals, for example, to address VCD in proteins and related compounds.

Pour en savoir plus, consultez le communiqué de presse associé à cet article : La quête des structures chirales supramoléculaires !
References:
Chiral Crystal Packing Induces Enhancement of Vibrational Circular Dichroism
Sascha Jähnigen, Arne Scherrer, Rodolphe Vuilleumier, and Daniel Sebastiani
Angew. Chem. Int. Ed. 2018, 57, 13344 –13348
DOI:10.1002/anie.201805671

Depuis 10 ans, le Programme L’Oréal-UNESCO Pour les Femmes et la Science a pour objectif de valoriser et accompagner des jeunes doctorantes et post-doctorantes à un moment charnière de leur carrière professionnelle. Ces dernières reçoivent une bourse, mais également des formations à la prise de parole en public et vulgarisation scientifique pour les aider à briser le plafond de verre.

Caroline ROSSI-GENDRON, qui a effectué son doctorat au sein du laboratoire PASTEUR du Département de Chimie de l’ENS, est lauréate de la Bourse L’Oréal-UNESCO Pour les Femmes et la Sciences 2018.

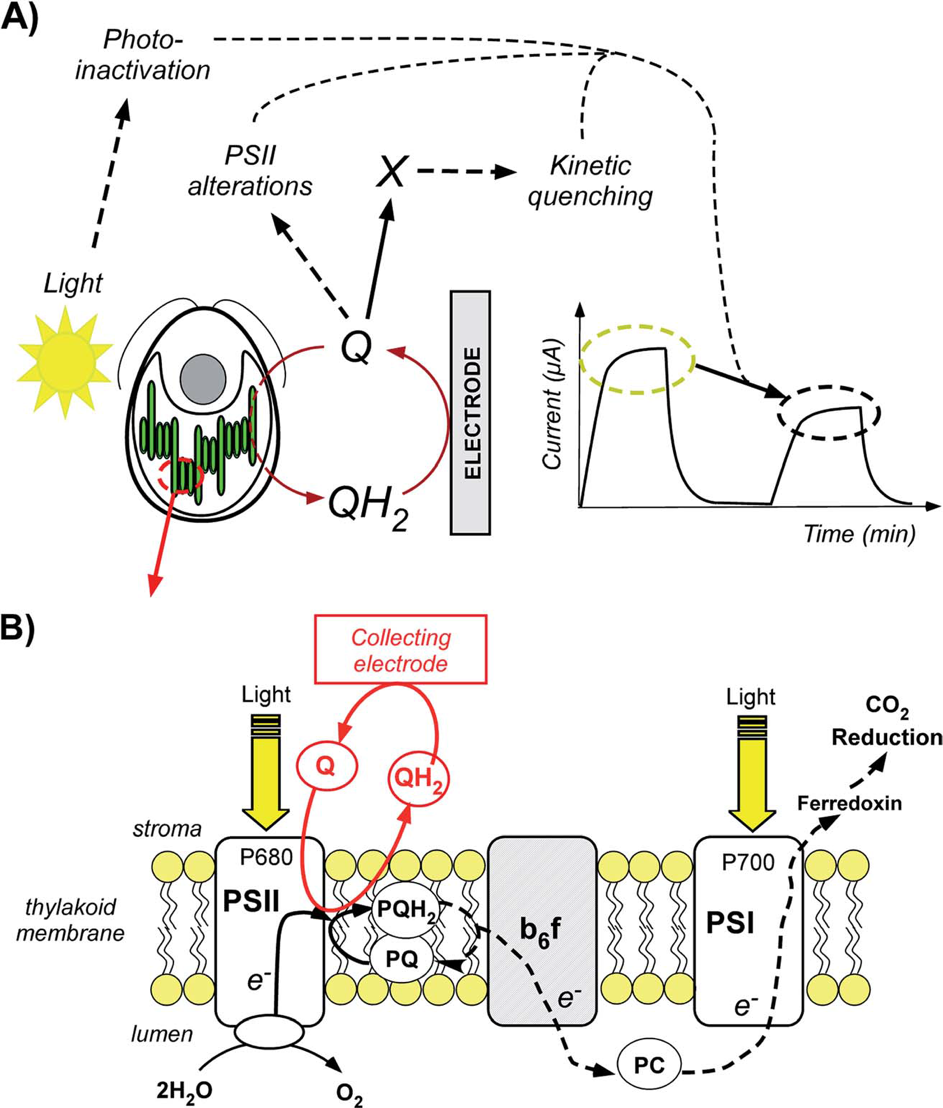
Plants, algae, and some bacteria convert solar energy into chemical energy by using photosynthesis. In light of the current energy environment, many research strategies try to benefit from photosynthesis in order to generate usable photobioelectricity. Among all the strategies developed for transferring electrons from the photosynthetic chain to an outer collecting electrode, we recently implemented a method on a preparative scale (high surface electrode) based on a Chlamydomonas reinhardtii green algae suspension in the presence of exogenous quinones as redox mediators. While giving rise to an interesting performance (10–60 μA cm−2) in the course of one hour, this device appears to cause a slow decrease of the recorded photocurrent. In this paper, we wish to analyze and understand this gradual fall in performance in order to limit this issue in future applications. We thus first show that this kind of degradation could be related to over-irradiation conditions or side-effects of quinones depending on experimental conditions. We therefore built an empirical model involving a kinetic quenching induced by incubation with quinones, which is globally consistent with the experimental data provided by fluorescence measurements achieved after dark incubation of algae in the presence of quinones.

Pour plus d’informations, consulter le communiqué de presse associé à cet article : Étudier la photoélectricité produite par des algues !
References:
Investigation of photocurrents resulting froma living unicellular algae suspension with quinones over time
Guillaume Longatte, Adnan Sayegh, Jérôme Delacotte, Fabrice Rappaport, Francis-André Wollman, Manon Guille-Collignon and Frédéric Lemaître
Chem. Sci., 2018
DOI: 10.1039/c8sc03058h

Thomas LE SAUX est lauréat du Prix de l’Instrumentation 2018 décerné conjointement par la Société Chimique de France et la Société Française de Physique.

Ce prix récompense une instrumentation innovante en Chimie Physique dont l’innovation porte sur un nouveau concept, une astuce technique, une nouvelle application… Les retombées de l’innovation pour la communauté scientifique et économique de la physicochimie seront un des critères d’évaluation.
Thomas LE SAUX a mis au point une instrumentation révolutionnaire développée pour la mise en œuvre de la modalité d’imagerie de fluorescence Speed OPIOM (Out-of-Phase Imaging after Optical Modulation). Speed OPIOM combine une modulation optimisée de l’illumination et une détection synchrone sensible à la phase qui permet de détecter sélectivement et quantitativement des sondes réversiblement photocommutables tout en s’affranchissant des contributions de fluorophores spectralement interférents ou de la lumière ambiante.
Compatible avec la microscopie, la macroscopie et l’endoscopie, Speed OPIOM ouvre la voie pour l’imagerie de fluorescence hautement multiplexée et bénéficiant d’un rapport signal sur bruit élevé, y compris en lumière du jour.